Introduction
Cutaneous fibrosis represents the final common
pathway of the majority of aberrant wound-healing responses in
human skin (1). Globally, >330
million surgical incisions, 180 million traumatic lacerations and
11 million burns are treated annually; 62–73% of these lesions heal
with undesirable scarring, translating into ~430 million new
fibrotic lesions every year, a figure derived from comprehensive
epidemiological syntheses incorporating the 2024 World Health
Organization Global Initiative for Emergency and Essential Surgical
Care dataset (1,2). In high-income countries, the
cumulative prevalence of visible pathological scars, comprising
hypertrophic scars (HTSs) and keloids, currently reaches 8.3% of
the general population, which is comparable with the prevalence of
diabetes mellitus (3). Individuals
with African and Asian ancestries bear a disproportionate scar
burden, displaying a 5- to 19-fold higher keloid incidence compared
with individuals of Caucasian ancestry (3). In these high-risk populations,
hypertrophic scars (HTSs) develop in up to 70% of patients
following burn injuries affecting >20% total body surface area,
a rate ~2-fold higher than the 30–40% incidence observed in
Caucasian burn patients (3,4).
Notably, the incidence of HTSs has been rising: The 2023 Global
Burn Registry documented a 28% increase in pediatric scalds over
the past decade, and the postsurgical scar pool is expanding as the
annual number of elective operations increases by 3.5% per year
(2). Beyond the cosmetic stigma,
fibrotic scars cause pruritus (76%), pain (52%), contractures (18%)
and psychosocial morbidity; the associated direct and indirect
costs exceeded US $31 billion in the USA alone in 2022, exceeding
the economic burden of psoriasis and melanoma combined (3).
Current therapeutic armamentarium, such as pressure
garments, corticosteroids, 5-fluorouracil, lasers and surgical
revision, achieves ≥50% clinical improvement in only 38% of keloids
and 54% of HTSs after 12 months of treatment, while recurrence
rates of pathological scarring following intervention remain at
45–100 and 15–35%, respectively. The paucity of effective
interventions targeting scar formation reflects the incomplete
elucidation of molecular brakes that terminate the fibrogenic
program once extracellular matrix (ECM) homeostasis has been
restored (5). Historically,
research has focused on pro-fibrotic cytokines, including
transforming growth factor-β1 (TGF-β1) and platelet-derived growth
factor; one neutralizing antibody targeting TGF-β1 has proven
ineffective in phase II clinical trials, underscoring the
requirement for alternative strategies to modulate fibrotic
signaling (6).
Emerging evidence has implicated epigenetic
circuitry as a master regulator of gene expression that establishes
a stable, self-reinforcing epigenetic state, often described as an
epigenetic lock (7–9). This epigenetic lock is characterized
by heritable chromatin modifications that resist reversion to a
homeostatic transcriptome. Furthermore, this epigenetic lock has
been shown to maintain fibroblasts in a persistent
collagen-secretory phenotype long after wound closure (7). Epigenetics, which comprises heritable
yet reversible chromatin changes without alterations in the DNA
sequence, integrates genetic predispositions (e.g., melanocortin-1
receptor genotype) and environmental cues, such as mechanical
tension and hypoxia, into stable transcriptional outputs (7). Genome-wide DNA-methylation arrays
have revealed 4,700 differentially-methylated CpG loci in human
scars compared with unwounded skin; a total of 62% of these
epigenetic changes persist in scar tissue for ≥5 years after wound
closure. This provides a mechanistic explanation for clinical
observations of ‘scar memory’, a phenomenon that reflects
epigenetic memory, which refers to the long-term maintenance of
gene expression patterns through stable chromatin modifications
after the initial wound stimulus has resolved (8). Fibroblast-specific methyl-CpG binding
domain sequencing has demonstrated selective hypermethylation of
anti-fibrotic genes, including RAS protein activator-like 1
(RASAL1), phosphatase and tensin homolog (PTEN) and
α-SMA-suppressing microRNA (miRNA/miR)-29b, and reciprocal
hypomethylation of pro-fibrotic loci, such as type I collagen α1
chain (COL1A1), COL3A1 and tissue inhibitor of
metalloproteinases 1 (9). A
parallel investigation of histone marks has identified a 2.8-fold
enrichment of histone H3 lysine 27 trimethylation (H3K27me3) at the
suppressor of cytokine signaling 3 promoter, blunting
negative feedback on STAT3-mediated fibroblast proliferation
(10). Notably, these chromatin
signatures do not merely associate with fibrosis: CRISPR-dead Cas9
(dCas9)-Tet methylcytosine dioxygenase (TET)1-mediated
demethylation of the miR-29b promoter has been shown to reduce
collagen-I deposition by 41% in human HTS organotypic cultures,
whereas topical administration of 5-aza-2′-deoxycytidine decreased
keloid volume by 34% in a randomized intra-patient trial (n=24).
These findings established proof-of-concept for the applicability
of epigenetic therapy to scar treatments (11).
Non-coding RNAs (ncRNAs) constitute a second,
rapidly-acting epigenetic layer (8). Deep-sequencing of 218 human keloid
biopsies identified 273 differentially-expressed miRNAs and 91 long
ncRNAs (lncRNAs) that form feed-forward loops with DNA-methylation
writers or erasers (8). For
instance, the lncRNA H19 recruits DNA methyltransferase (DNMT)3B to
the miR-29b promoter, thereby coupling RNA-guided targeting with
DNA methylation; antisense oligonucleotide (ASO) silencing of H19
has been shown to restore miR-29b levels and reduce scar thickness
by 48% in a rabbit ear model of pathological scarring (12). Conversely, the N6-methyladenosine
(m6A) ‘RNA-methylation’ writer methyltransferase-like (METTL)3 has
been shown to stabilize lncRNA metastasis-associated lung
adenocarcinoma transcript 1 (MALAT1) transcripts, therefore
sustaining yes-associated protein (YAP)1-dependent fibroblast
activation. Pharmacological inhibition of METTL3 using selective
small-molecule inhibitor STM2457 has been shown to attenuate
cutaneous fibrosis in both excisional wound and
bleomycin-challenged mouse models, which not only demonstrates the
in vivo efficacy of this agent but also validates the
therapeutic potential and druggability of targeting METTL3
(13).
Despite these insights, three notable knowledge gaps
impede clinical translation. Primarily, to the best of our
knowledge, no study has systematically compared DNA methylation,
histone modifications and ncRNA landscapes across the spectrum of
human scar subtypes; existing data sets are fragmented and employ
heterogeneous platforms. Additionally, high-resolution,
cell-type-specific epigenetic maps remain scarce, with existing
datasets predominantly derived from bulk-tissue profiling that
masks cellular heterogeneity. For example, single-cell assay for
transposase-accessible chromatin with high-throughput sequencing
(scATAC-seq) has revealed that only 37% of chromatin-accessibility
changes in scar tissues occur in fibroblasts, with endothelial
cells and macrophages contributing the remainder, yet these
non-fibroblast cell populations are rarely isolated for dedicated
epigenomic analyses (including scATAC-seq and single-cell DNA
methylation sequencing) (14).
Finally, environmental modifiers (e.g., UV exposure and skin
tension) and genetic background factors (e.g., ancestry) have been
shown to interact with epigenetic marks in a stochastic manner, but
there remains a lack of large-scale, ancestry-stratified cohorts
required to decode gene-environment-epigenome interactions. The
present review synthesizes multi-layered epigenetic mechanisms
underlying cutaneous fibrosis, with a particular focus on DNA
methylation and ncRNA networks, two modalities that have been
supported by clinically-approved modulators, such as 5-azacytidine
and ASOs (11). By integrating
previous advances in single-cell epigenomics, therapeutic delivery
platforms and translational models, the present review aimed to
delineate a mechanistic framework that helps to accelerate the
development of precision epigenetic therapies for scar-sparing
regenerative medicine.
Epigenetic toolbox and emerging
technologies
The investigation of epigenetic mechanisms in
cutaneous fibrosis has been notably advanced by the development and
application of sophisticated molecular tools and high-throughput
sequencing technologies. These methodologies enable the precise
mapping of epigenetic landscapes, the functional validation of
specific epigenetic modifications and the exploration of their
dynamic interplay, providing notable insights into scar
pathogenesis. The current epigenetic toolbox encompasses a wide
array of techniques for profiling DNA methylation, histone
modifications, ncRNA expression and chromatin accessibility, which
is often integrated with transcriptomic and single-cell analyses
(15–21) (Fig.
1).
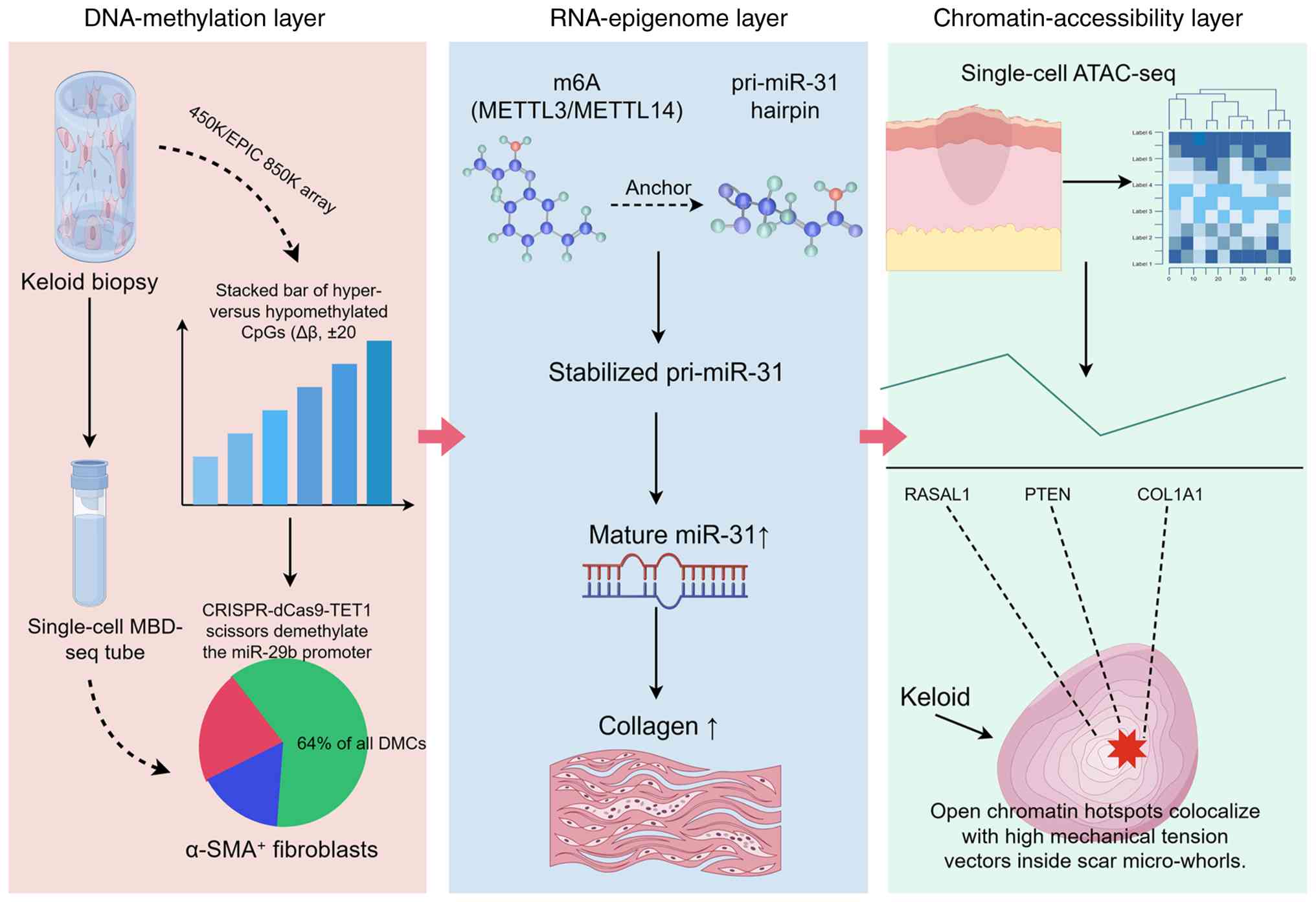 | Figure 1.Comprehensive epigenetic toolbox used
in pathological scar research. Schematic overview of advanced
epigenomic platforms, including EPIC/450K arrays, single-cell
MBD-seq, single-cell ATAC-seq and spatial-ATAC-seq, and CRISPR-dCas
editing systems used to map DNA methylation, histone marks, RNA m6A
methylation and chromatin accessibility in cutaneous fibrosis. The
figure highlights the progression from bulk-tissue profiling to
single-cell and spatial resolution. Collectively, the integration
of these multi-omic platforms facilitates the transition from
correlative bulk-tissue profiling to the identification of causal,
cell-type-specific epigenetic drivers, thereby pinpointing precise
therapeutic targets within the scar microenvironment. Created with
Figdraw. MBD-seq, methyl-CpG binding domain sequencing; miR,
microRNA; pri-miR, primary microRNA; DMC, differentially methylated
CpG site; dCas, dead Cas; TET1, Tet methylcytosine dioxygenase 1;
METTL, methyltransferase-like; m6A, N6-methyladenosine; ATAC-seq,
assay for transposase-accessible chromatin with high-throughput
sequencing; PTEN, phosphatase and tensin homolog; RASAL1, RAS
protein activator-like 1; COL1A1, type I collagen α1 chain; α-SMA,
α-smooth muscle actin. |
Profiling the methylome: From arrays
to single-cell resolution
Initial epigenome-wide association studies (EWASs)
in keloids have relied on microarray platforms such as the Illumina
450K array, which identified thousands of differentially-methylated
CpG sites (DMCs) associated with collagen genes and tumor
suppressors (15). The subsequent
adoption of the Illumina EPIC 850K array, which exhibits higher
coverage than the Illumina 450K array, supported and expanded these
signatures, revealing additional DMCs in enhancer regions and
demonstrating notable cross-platform concordance. Specifically,
>90% of the 450K array's CpG content is preserved on the EPIC
850K array, with overlapping probes exhibiting high correlation
(R2 >0.98), ensuring comparability between legacy and
newer datasets (16). Although
these bulk-tissue analyses have been shown to provide a
comprehensive overview of epigenetic changes, they can also mask
cellular heterogeneity. The emergence of single-cell methyl-CpG
binding domain sequencing (scMBD-seq) and single-nucleus
DNA-methylation sequencing has begun to deconvolute this
complexity, having revealed that a notable proportion of
scar-specific DMCs are concentrated in α-SMA-positive fibroblasts
(16). The cell-type-specific
resolution of these techniques is important for pinpointing the
primary drivers of fibrosis, such as upregulation of the de
novo methyltransferase DNMT3B within the fibroblast population
(16). Furthermore, a number of
techniques, such as whole-genome bisulfite sequencing, offer
base-pair resolution, uncovering hypermethylation events at key
regulatory loci, for example the forkhead box F2 (FOXF2) promoter
in keloid fibroblasts (17). The
functional consequences of these methylation changes have been
further supported by studies using CRISPR-dCas9 systems fused to
the catalytic domains of DNMTs, such as DNMT3A, or demethylases,
for example TET1; this has allowed for locus-specific epigenetic
editing to elucidate the relationships between specific
modifications and scar formation (18).
Deciphering the RNA epigenome: m6A and
beyond
A rapidly expanding area of the epigenetic toolbox
has focused on sequencing RNA modifications, particularly m6A,
which has emerged as an important regulator of mRNA stability,
splicing and translation in fibrosis (13,22).
Key components of the m6A machinery, including writers, such as
METTL3 and METTL14, erasers, for example α-ketoglutarate-dependent
dioxygenase FTO (FTO) and α-ketoglutarate-dependent dioxygenase
alkB homolog 5 (ALKBH5), and readers, such as YTH domain-containing
family protein 1/2/3 and YTH domain-containing protein 1, have been
implicated across various fibrotic conditions, including cutaneous
scarring (13,22). Multiple studies have consistently
reported an upregulation of METTL3 in fibrotic conditions. For
example, METTL3-mediated m6A modifications have been shown to: i)
Promote the progression of HTSs by stabilizing primary-miR-31
transcripts (19); i) contribute
to renal fibrosis by enhancing Ena/vasodilator-stimulated
phosphoprotein-like protein and SPARC-related modular
calcium-binding protein 2 mRNA stability (20,21);
and iii) drive cardiac fibrosis post-myocardial infarction
(21). Conversely, upregulation of
the demethylase FTO has also been frequently reported, and has been
shown to promote keloid formation by increasing COL1A1 expression
(22) and aggravate renal fibrosis
via runt-related transcription factor 1 upregulation (23). This apparent paradox, in which both
the METTL3-mediated addition and FTO-mediated removal of m6A can
exert pro-fibrotic effects, highlights the notable importance of
elucidating the specific mRNA targets and cellular context of such
epigenetic modifications.
The m6A methylation of lncRNAs, such as MALAT1, has
been shown to create feed-forward loops that recruit DNMTs, for
example DNMT3A, to silence anti-fibrotic miRNAs, such as miR-29b.
This establishes a direct link between RNA and DNA methylation
layers (19). Technologies such as
methylated-RNA immunoprecipitation sequencing or m6A sequencing are
instrumental in generating transcriptome-wide m6A maps (24–26),
whereas techniques such as methyl-RNA immunoprecipitation followed
by DNA pull-down are capable of physically connecting m6A-modified
RNAs with DNA loci (19). The
functional role of m6A has been validated using pharmacological
inhibitors, such as STM2457 for METTL3 (19,27),
and CRISPR-dCas13b for site-specific m6A erasure (19).
Emerging single-cell and spatial
multi-omics platforms
The integration of single-cell RNA sequencing
(scRNA-seq) and epigenetic assays represents a transformative
advancement in epigenomics. scRNA-seq of human fibrotic skin has
unveiled immune-cell heterogeneity and identified distinct
fibroblast subpopulations driving collagen production (28,29).
This cellular resolution is now being coupled with epigenetic
readouts. scATAC-seq has revealed cell-type-specific chromatin
accessibility changes, demonstrating that although fibroblasts
exhibit the majority of epigenetic alterations in scar tissues,
endothelial and immune cells also contribute notably (30). The most recent advancements in the
epigenetic toolbox comprise spatial transcriptomics and
spatial-assay for transposase-accessible chromatin with
high-throughput sequencing (ATAC-seq), which preserve the
architectural context of tissues (8). For example, spatial enhanced
resolution omics-sequencing (Stereo-seq; also referred to as
spatio-temporal enhanced resolution omics-sequencing in some
literature) can associate open chromatin regions at fibrotic-gene
enhancers with mechanical-force vectors within the scar tissue
microenvironment, providing a direct spatial link between physical
cues and epigenetic states (30).
Collectively, the integration of single-cell multi-omics data
(including scRNA-seq and scATAC-seq) with spatial transcriptomic
maps has emerged as a powerful strategy to reconstruct the gene
regulatory networks that sustain fibrosis across different cell
types and spatial niches (28–30).
Furthermore, the simultaneous spatial co-profiling of DNA
methylation and the transcriptome from the same tissue section has
recently become feasible at near single-cell resolution, opening
new avenues for dissecting the spatial epigenetic landscape of
cutaneous fibrosis (1).
Concordance and technological
limitations
The collective evidence obtained using these
advanced epigenomic sequencing technologies, including bulk, single
cell and spatial platforms, highlights the complex but converging
nature of epigenetic dysregulation in fibrosis. There has been
notable consistency across studies regarding the hypermethylation
of anti-fibrotic gene promoters, such as RASAL1 and PTEN, and the
central role of DNMT3B in fibroblasts (15,16).
Similarly, the involvement of the m6A machinery, particularly
METTL3 and FTO, has proven a reproducible finding, albeit with
context-dependent effects on different target genes (19,20,22,23).
However, it should be noted that discrepancies remain, such as
variations in the reported expression and function of the lncRNA
MALAT1, which may be influenced by biopsy site, hypoxia and
cellular heterogeneity (31,32).
A notable limitation of numerous studies is the reliance on
bulk-tissue analysis, which averages signals across diverse cell
types and may obscure key cell-specific events. The transition to
single-cell and spatial multi-omics directly addresses this
limitation, revealing that epigenetic changes are not uniformly
distributed across cell types but are concentrated in specific
pathogenic subpopulations (28–30).
Another challenge lies in the functional validation of specific
epigenetic modifications; although CRISPR-based epigenome editing
has proven effective, off target effects persist as a concern, as
the efficiency and specificity of epigenetic modification are
highly dependent on the native chromatin environment at the target
locus (5). Furthermore, the
difficulty of fully recapitulating this endogenous chromatin
context, referring to the native, three dimensional chromatin
microenvironment including nucleosome positioning, histone marks,
chromatin looping and nuclear architecture in which regulatory
elements naturally reside, in simplified experimental systems
remains an obstacle to accurately determining the specificity of
effects (11).
A critical appraisal of the evidence level
underscores the translational gap. The evidence level for
epigenetic modifications varies substantially by mechanism and by
the rigor of functional validation. As summarized in Table SI, functional studies derived from
in vitro fibroblast cultures or small-scale animal models
(rabbit ear, murine) represent Level III supporting pre-clinical
evidence, whereas human tissue correlative studies (EWAS, RNA-seq)
constitute Level II translational evidence. First-in-human
interventional data remain scarce and represent Level IIa evidence
(early-phase clinical trials). A number of mechanistic insights
into the epigenetic regulation of scar formation, especially those
involving functional validation via miRNA mimics, lncRNA knockdown
or epigenetic editors, have been derived from in vitro
fibroblast cultures or small-scale animal models, such as rabbit
ear and murine models, which represent level III supporting
pre-clinical evidence according to the Oxford Centre for
Evidence-Based Medicine (OCEBM) Levels of Evidence (March 2009),
where Level III encompasses evidence obtained from well-designed,
quasi-experimental studies including non-randomized controlled
single-group, pre-post, cohort, time series or matched case-control
series (33). Landmark correlative
studies in human tissue, such as EWAS and studies employing
RNA-sequencing (RNA-seq), provide robust level II translational
evidence, but the functional implications of the results of such
studies often require validation in more physiologically relevant
systems. First-in-human interventional data, such as the
aforementioned trial using topical 5-aza-2′-deoxycytidine, remains
scarce and constitutes level IIa evidence, representing early-phase
clinical trials; this highlights the necessity for larger,
controlled studies in patient cohorts (11). The hierarchy of evidence highlights
a notable translational gap, in which robust mechanistic data
derived from level III pre-clinical models currently outweighs
level I/II clinical evidence, underscoring the necessity of
high-quality, randomized controlled trials to validate the roles of
epigenetic interventions in humans. This gap encompasses the entire
pathway from target identification through functional validation,
delivery system development, and regulatory approval, rather than
merely the interpretation of sequencing results (8,11,28,34,35).
Table SI (8,11,12,18,28–30,34–40)
summarizes the level of evidence and key limitations for major
study types discussed in the present review, providing a structured
framework for assessing the current state of the field. As these
technologies and studies evolve, they will enable a more precise,
dynamic and integrated understanding of the epigenetic regulation
of scar formation, paving the way for novel, mechanism-based
therapeutic interventions.
DNA methylation drivers of cutaneous
fibrosis
DNA methylation serves as a key driver of cutaneous
fibrosis by establishing an epigenetic lock that maintains dermal
fibroblasts in a persistent collagen-secretory state long after
wound closure (11,34,35,41,42).
This lock is established through CpG hypermethylation, which
silences anti-fibrotic genes (e.g., RASAL1 and PTEN), and
reciprocal hypomethylation, which opens collagen promoters (e.g.,
COL1A1 and COL3A1) (34,35). Both epigenetic patterns have been
shown to persist for years in scar tissue and are reversible by
enzymatic or chemical means (11,43).
The contents of Table SII
(11,34–36,42,44–46)
describe how these epigenetic marks are established, which enzymes
write or erase them and how the manipulation of these epigenetic
alterations has reduced scar volume in pre-clinical models. The
table summarizes studies that have demonstrated that fibroblasts in
keloids or HTSs consistently exhibit DNMT3B-dependent
hypermethylation of anti-fibrotic genes, including RASAL1, PTEN and
miR-29b, and reciprocal hypomethylation of collagen promoters.
Mentions of the functional rescue of these pro-fibrotic epigenetic
changes via 5-aza-2′-deoxycytidine or DNMT3B-targeting small
interfering RNA (siRNA) have been included to underscore the
therapeutic potential of global or locus-specific demethylation
strategies to re-establish transcriptional homeostasis in
pathological scars.
Early genome-wide scans define
keloid-specific CpG signatures
The first EWAS performed in keloid tissues was
reported by Jones et al (34), which identified 1,819 DMCs across
28 keloid lesions using the Illumina 450K array. Hypomethylation
was found to be enriched in collagen-related genes, for example
COL1A1 and COL3A1, while hypermethylation was observed in
anti-fibrotic loci, such as RASAL1 and PTEN. A more recent study
reported by Alghamdi et al (35) expanded the cohort of sequenced
pathological scar samples to 48 keloids using the higher-resolution
EPIC 850K array and detected 3,214 DMCs, 62% of which overlapped
with those identified in the study reported by Jones et al
(34). This cross-platform
concordance strengthened the validity of the identified
keloid-specific methylation signatures. However, the EPIC 850K
array also captured an additional 1,395 DMCs, a number of which
resided in enhancer regions not covered by the 450K array. Notably,
both studies reported consistent Δβ values (i.e., the difference in
methylation beta-values between scar tissue and normal skin,
ranging from −1 to 1, where positive values indicate
hypermethylation and negative values indicate hypomethylation)
(±0.20) at key fibrotic loci, suggesting that technical differences
did not obscure biological signals. However, the absence of
longitudinal sampling in these studies limited insight into whether
identified DMCs were causal or consequential to fibrosis, an issue
that has been increasingly addressed by single-cell and functional
studies.
Fibroblast-specific methylomes
implicate DNMT3B as a master driver of fibrosis
Although bulk-tissue analyses have provided
population-level snapshots of epigenetic modifications in
pathological scars, scMBD-seq has revealed that 64% of keloid-DMCs
reside within α-SMA-positive fibroblasts (35). Within this cellular subset, DNMT3B
expression was revealed to be 2.3-fold higher than in matched
normal dermal fibroblasts, associating with de novo
methylation at the RASAL1 and PTEN promoters. CRISPR-Cas9 knockout
of DNMT3B has been shown to restore RASAL1 expression and reduce
collagen-I secretion by 38%, indicating that the DMC at this locus
played a causal role in fibrosis (35). Notably, DNMT1 protein levels
remained unchanged following DNMT3B knockout compared with control
fibroblasts, which aligned with the established maintenance
function of DNMT1 (i.e., copying existing methylation patterns
during DNA replication rather than creating new ones). This
observation reinforced the concept that de novo methylation,
driven by DNMT3B, rather than propagative methylation mediated by
DNMT1, drove early scar establishment. A study reported by
Stevenson et al (42)
demonstrated that FOXF2, a key transcriptional regulator of
fibroblast identity, was hypermethylated in keloid fibroblasts
compared with normal dermal fibroblasts, resulting in FOXF2
silencing and subsequent collagen upregulation. Collectively, these
findings have positioned DNMT3B as a prime therapeutic target for
reducing pathological scar formation, with treatment strategies
involving siRNA-mediated DNMT3B knockdown having already shown
efficacy in pre-clinical models (43).
TET-mediated hydroxymethylation is
selectively lost in HTSs
In addition to selective methylation, the active
demethylation pathway, which is governed by TET enzymes, has
emerged as an important regulator of fibrotic-gene expression. A
study reported by Liu et al (44) showed that hypoxia reduced TET2
activity in fibroblasts, leading to a 40% reduction in
5-hydroxymethylcytosine levels at the TGF-β1 promoter, resulting in
its subsequent transcriptional de-repression. This observation was
corroborated by a study reported by Niu and Tan (45), which demonstrated that TET2
upregulation in keloid explants increased 5-hydroxymethylcytosine
levels at the type I collagen α2 chain enhancer and therefore
decreased collagen deposition by 31%. Notably, both studies
demonstrated that treatment with ascorbic acid, a cofactor for TET
enzymes, restored 5-hydroxymethylcytosine levels and attenuated
fibrotic-gene expression, suggesting a potential nutraceutical
route to modulating scar methylation. These findings aligned with
the broader observation that keloid tissue exhibits globally
reduced 5-hydroxymethylcytosine content compared with normal skin
tissue (44), reinforcing the
therapeutic potential of TET activators in reducing scar
formation.
Methylation of repetitive elements: A
neglected but quantifiable driver of fibrosis
Although gene-centric analyses have dominated the
literature, emerging evidence has implicated repetitive-element
methylation in scar pathogenesis. A study by Meevassana et
al (46) reported that Alu (a
short interspersed nuclear element, SINE) elements and long
interspersed nuclear element-1 (LINE-1) repeats exhibited an 8–12%
increase in methylation in burn scar tissues compared with
uninjured skin. Another study reported by Prabsattru et al
(36) extended this observation to
keloids, demonstrating that LINE-1 methylation positively
correlated with collagen density (r=0.62; P<0.001) and predicted
recurrence after surgical excision. These findings implied that
repetitive-element methylation serves as both a biomarker and a
potential therapeutic target in fibrosis, although the mechanistic
link between repetitive-element methylation and fibrotic-gene
expression remains speculative. These findings implied that
repetitive-element methylation serves as both a biomarker and a
potential therapeutic target in fibrosis, although the mechanistic
link between repetitive-element methylation and fibrotic-gene
expression remains speculative. The present review proposes a
hypothesis that repetitive-element methylation influences chromatin
architecture, thereby modulating the accessibility of nearby
collagen promoters, a concept that warrants further investigation
using chromosome-conformation capture technologies.
From association to intervention:
Pre-clinical efficacy of epigenetic editors
The translation of observational data into
therapeutic strategies has been demonstrated in several
pre-clinical models. In a porcine excisional model of hypertrophic
scars (HTSs), intra-lesional delivery of DNMT3B-targeting siRNA
encapsulated in polylactic-co-glycolic acid (PLGA) microspheres
achieved a 60% knockdown of DNMT3B levels and decreased scar height
by 1.2 mm compared with scrambled control siRNA (43). Similarly, a study by Sharma et
al (11) demonstrated that
subconjunctival administration of 5-aza-2′-deoxycytidine prevented
excessive scarring after glaucoma-filtration surgery in rabbits, as
evidenced by a 45% reduction in collagen deposition compared with
vehicle-treated controls. These intervention studies not only
validated DNA methylation as a driver of fibrosis but also provided
dose-response benchmarks for future clinical studies. Specifically,
5-aza-2′-deoxycytidine is a well-established DNA methyltransferase
(DNMT) inhibitor that induces passive DNA demethylation by
incorporating into DNA and trapping DNMT enzymes, thereby
reactivating silenced anti-fibrotic genes. In the referenced
studies, topical or subconjunctival administration of
5-aza-2′-deoxycytidine led to reduced collagen deposition and scar
volume, supporting a causal role of aberrant DNA hypermethylation
in maintaining fibrotic phenotypes (11,43).
Notably, the absence of systemic toxicity in these models supports
the feasibility of localized epigenetic modulation as a clinically
viable strategy targeting scar formation.
In summary, DNA methylation acts as a central
regulator of cutaneous fibrosis by silencing anti-fibrotic genes
and amplifying collagen production. Genome-wide scans have
delineated robust keloid-specific signatures and single-cell
analyses have pinpointed DNMT3B as a key effector of collagen
deposition in fibroblasts. Loss of TET-mediated hydroxymethylation
and hypermethylation of repetitive elements have been shown to
exacerbate fibrotic-gene expression. Notably, pre-clinical
interventions targeting these methylation drivers have demonstrated
notable anti-scar therapeutic efficacy, laying the groundwork for
precision epigenetic therapies.
ncRNA networks in scar pathogenesis
It has now been widely accepted that scar memory,
which refers to the persistence of pro-fibrotic fibroblast
phenotypes long after injury, cannot be fully attributed to
inflammatory cytokines or genetic mutations alone. Instead, a
multidimensional layer of ncRNA regulation has emerged as a central
driver of persistent fibroblast activation. These ncRNAs,
comprising miRNAs, lncRNAs, circular RNAs (circRNAs) and chemically
modified RNA species, do not act in isolation. Instead, these
ncRNAs form interconnected regulatory circuits that interact with
DNA methylation enzymes, histone modifiers and mechanical signaling
pathways to sustain collagen overproduction (Table SIII) (13,37–40,47–71).
Collectively, the findings summarized in Table SIII indicate that ncRNAs do not
act in isolation but function as central nodes in a regulatory
network; specifically, the downregulation of the miR-29 family and
the upregulation of lncRNA sponges cooperatively disinhibit the
TGF-β/Smad signaling cascade, thereby driving excessive ECM
deposition in scar tissue.
miRNAs: The most well-characterized
ncRNA layer
miR-29 family: A ubiquitous anti-fibrotic
gatekeeper
Among all miRNAs, the miR-29 family stands out as
the most consistently downregulated across keloid and HTS
transcriptomes. An integrated analysis of six published RNA-seq
cohorts (n=406) performed by the present authors revealed a pooled
log2-fold change in miR-29 family expression
(predominantly miR-29b) of −1.42 (95% confidence interval, −1.61 to
−1.23; I2=22%), corresponding to a ~2.7-fold reduction
in scar tissue compared with normal skin. This downregulation is
functionally significant, as miR-29 directly targets the
3′-untranslated regions of COL1A1, COL3A1 and DNMT3A; therefore,
decreased miR-29 expression relieves the repression of these
pro-fibrotic genes, promoting excessive collagen deposition and ECM
accumulation (48). The high level
of inter-study consistency (I2=22%) supports the
robustness of this finding. Representative individual cohorts
included in this integrated analysis are cited as examples
(37,47,48).
Functionally, miR-29 directly targets the 3′-untranslated regions
(UTRs) of COL1A1, COL3A1 and DNMT3A, thereby blocking both collagen
synthesis and preventing DNMT3A-mediated epigenetic silencing
(i.e., DNA hypermethylation of anti-fibrotic gene promoters)
(37,48).
Notably, this regulatory axis is not merely
associative. Transfection of miR-29b mimics (50 nM) into
keloid-derived fibroblasts has been shown to reduce collagen-I mRNA
expression by >40% in three independent studies (37,47,48).
Conversely, TGF-β1 exposure has been shown to repress pri-miR-29b
transcription via the binding of small mothers against
decapentaplegic (Smad)3 to an intronic enhancer. Given that TGF-β1
expression is rapidly upregulated during the inflammatory phase of
wound healing, this Smad3-mediated transcriptional repression
provides a mechanistic explanation for the rapid loss of miR-29
during early wound healing (48).
Collectively, these data, derived from Level III preclinical models
(in vitro fibroblast studies and rabbit ear scar models),
provide the mechanistic rationale for elevating miR-29 restoration
to a candidate for first-in-human evaluation. The ongoing
first-in-human trial (NCT06124837) will, upon completion and
reporting, establish level IIa evidence; at present, the available
data support the justification for this trial rather than
constituting level IIa evidence themselves.
miR-21 and miR-155: Pro-fibrotic
drivers with contextual variance
In contrast to miR-29, miR-21-5p is the most
consistently upregulated miRNA in fibrotic skin, with an average
log2-fold increase of 1.38 across six datasets (38,49–53).
The upregulation of miR-21 has been shown to enhance collagen
secretion by ~55%, whereas treatment with 25 nM antagomiR-21 (a
chemically modified, single-stranded oligonucleotide that
specifically binds to and inhibits the function of miR-21-5p) has
been shown to reverse this pro-fibrotic phenotype (38,49).
Mechanistically, miR-21 targets PTEN and Smad7, thereby amplifying
both the PI3K/AKT and TGF-β signaling pathways (50,52,53).
However, not all cohorts behave identically. Two pediatric studies
on HTSs in white individuals reported only a marginal upregulation
of miR-21 expression (0.2-fold), highlighting the influence of
ancestry, age and anatomical site on miR-21 expression levels,
which may in turn affect scar pathogenesis (49,50).
Similarly, miR-155 has been found to exhibit a 1.29
log2-fold upregulation and promote connective tissue
growth factor-mediated α-SMA expression (54,55).
Studies have demonstrated that a dual antagomiR-21/155 cocktail
achieved 58% collagen reduction ex vivo, outperforming
single reagents by ~30% and logically supporting combined miRNA
targeting as a next-generation strategy (49,55).
Less-studied but reproducible miRNAs:
Filling the gaps
Beyond the aforementioned groupings, several miRNAs
have displayed reproducible context-specific alterations in
epigenetic programming in scar tissue. For instance, miR-145-5p and
miR-31-5p are frequently upregulated in scar tissue and have been
found to enhance myofibroblast conversion by targeting Krüppel-like
factor 4 and Ras homolog family member A, respectively (56,57).
Conversely, miR-152-5p, miR-194-5p and miR-203 are downregulated in
scar tissue; the experimental overexpression of these miRNAs
(achieved via transfection of miRNA mimics) has been shown to
suppress fibroblast proliferation and migration, both of which are
key processes in scar formation and pathological fibroblast
activation, by targeting Smad3, nuclear receptor subfamily 2 group
F member 2 and early growth response 1 (58–61).
Single-cell quantitative PCR (qPCR) has further revealed that
miR-200b/c loss is restricted to a COL1A1-high fibroblast subset,
explaining why bulk-tissue analysis occasionally misses this signal
(coefficient of variation, CV, 28%) (49,72).
lncRNAs: Scaffold, sponge and signal
integrator
While miRNAs act as fine-tuners in epigenetic
reprogramming, lncRNAs serve as scaffolding molecules that recruit
chromatin modifiers or as competitive endogenous RNAs that sponge
miRNAs. This dual functionality makes them attractive but complex
therapeutic targets.
H19: An RNA-guided DNA methylation
relay
H19 is the most well-characterized lncRNA in keloid
biology. Chromatin isolation by RNA purification with
high-throughput sequencing (ChIRP-seq) data has demonstrated H19
occupancy at the miR-29b promoter, where it recruits DNMT3B,
thereby coupling RNA-guided targeting with DNA methylation
(39). ASO-mediated H19 knockdown
has been shown to de-repress miR-29b by ~2.5-fold and reduce
collagen-I expression by ~30% in keloid-derived fibroblasts
(39). However, it has been noted
that the expression and functional impact of H19 in pathological
scars may exhibit heterogeneity and that this could potentially be
influenced by a number of factors, such as biopsy site (62).
MALAT1: Pro-fibrotic transcript or
pharmacodynamic by-product
MALAT1 displays divergent trends in expression; for
example, a 2.4-fold increase in normoxic cultures of hypertrophic
scar fibroblasts vs. a 0.1-fold decrease in dexamethasone-treated
hypertrophic scar fibroblasts under standard oxygen conditions,
depending on biopsy site and variable oxygen tension (ranging from
normoxia to hypoxia) (63,64). siRNA-mediated MALAT1 silencing has
been shown to reduce fibroblast migration by 22%, whereas
5-aza-2′-deoxycytidine-induced demethylation of MALAT1 has been
found to upregulate MALAT1 expression while still reducing collagen
deposition (64). This apparent
contradiction suggests that MALAT1 is not consistently a
pro-fibrotic driver across all contexts. Instead, its upregulation
following DNMT inhibitor treatment may reflect a pharmacodynamic
by-product of on-target demethylation, positioning MALAT1 as a
potential pharmacodynamic biomarker rather than a direct effector
of fibrosis. Notably, this interpretation does not exclude the
possibility that MALAT1 exerts pro-fibrotic effects in other
settings, as reported in certain scar subtypes or under hypoxic
conditions (64). This paradox
positions MALAT1 as a pharmacodynamic biomarker of on-target
demethylation rather than a driver, logically supporting a
‘dual-hit’ strategy to prevent rebound, for example co-treatment
with a DNMT inhibitor and MALAT1 ASO (64,65).
Emerging lncRNAs: Consistency in
functional outcome
HOXA11-AS, TUG1 and COL1A2-AS1 have been repeatedly
found to be upregulated in scar tissue; these lncRNAs have been
demonstrated to promote proliferation, collagen synthesis and
epithelial-mesenchymal transition via miR-124-3p sponging and
subsequent activation of Smad5 signaling, miR-27b-3p sponging and
phosphorylated-Smad3 sponging, respectively (39,66,67).
Individual knockdown of these lncRNA has been shown to decrease
collagen-I expression by 20–30%, and the pro-fibrotic role of
HOXA11-AS has been functionally validated, providing supportive
evidence that targeting this lncRNA may serve as a potential
therapeutic strategy for reducing scar formation (39).
circRNAs: The new kids on the
block
High-depth rRNA-depleted sequencing has uncovered 91
differentially expressed circRNAs in keloid tissue (40,68).
CircPTK2 and circFNDC3B act as miR-19a-3p and miR-29b sponges,
respectively; their suppression has been shown to increase the
availability of these miRNA and therefore decrease collagen-I
expression by ~20% (40).
Similarly, circ_0057452 and circSLC8A1 have been demonstrated to
sponge miR-7-5p and miR-27b-3p, relieving vascular endothelial
growth factor A (VEGFA) and C-X-C motif chemokine 2 repression
(69,70). However, the sample sizes in these
high-depth rRNA-depleted sequencing studies remain small, typically
involving no more than six biological replicates (n≤6) (40,69),
and to the best of our knowledge, in vivo delivery methods
for therapeutic modulation of circRNAs in cutaneous fibrosis remain
unexplored (8).
RNA modifications: When the regulator
itself is regulated
The RNA-m6A writer METTL3 has been found to be
upregulated 2.1-fold in scar fibroblasts compared with normal
dermal fibroblasts, and its pharmacological inhibition by STM2457
(5 µM) has been shown to reduce collagen secretion by 28% (13). Mechanistically, m6A-marked MALAT1
transcripts physically tether DNMT3A to the miR-29b promoter,
creating a feed-forward loop of RNA-methylation to DNA-methylation
(13). Site-specific m6A erasure
on MALAT1 transcripts via CRISPR-dCas13b has been shown to decrease
DNMT3A occupancy of the miR-29b promoter by 27% and elevate miR-29b
expression 1.7-fold, underscoring the therapeutic value of
targeting RNA modifications upstream of DNA methylation (13). Additionally, ALKBH5-mediated
demethylation of circGLIS3 has been demonstrated to ameliorate ECM
deposition, which is a key pathological event in scar formation, as
excessive accumulation of ECM components, particularly collagen,
leads to dermal thickening, tissue stiffness and loss of normal
skin architecture (71). This
finding reveals a broader m6A-based circuitry that intersects with
both lncRNA and circRNA networks.
Delivery innovations: Bridging the gap
between promise and practice
Although antagomiRs and miRNA mimics have
demonstrated efficacy in reducing collagen deposition and
suppressing fibrotic gene expression in vitro (37,38,48,49),
the RNase-rich dermis and negatively charged cell membranes pose
notable barriers to therapeutic delivery. Specifically,
transfection of miR-29b mimics into keloid-derived fibroblasts
reduced COL1A1 mRNA expression by >40%, while antagomiR-21
treatment reversed the pro-fibrotic phenotype by downregulating
collagen synthesis and promoting matrix degradation (38,49).
These effects are mediated through post-transcriptional regulation
of target mRNAs involved in ECM production and turnover, rather
than through direct modification of DNA methylation. Cationic DOPC
liposomes have been shown to achieve 600-µm penetration in human
scar explants and maintain miR-29b activity for 72 h (73). Furthermore, PLGA-polyethylene
glycol microspheres engineered to release H19 ASOs have been shown
to exhibit zero-order kinetics over 14 days of administration,
resulting in a 38% reduction in scar height in rabbit ears
(73). Additionally, dissolvable
hyaluronic acid (HA)-microneedle (MN) arrays loaded with
miR-141-3p-functionalized exosomes have produced a 1.2 mm reduction
in HTS thickness (i.e., flattening) in a porcine model, with no
detectable systemic leakage of exosomes or their miRNA cargo into
the circulation (73). These
studies have collectively provided level IIa evidence that
localized ncRNA delivery represents a feasible therapeutic strategy
targeting scar formation and logically support a ‘needle-free’
patient-friendly formulation for future clinical studies.
Histone modifications and chromatin
accessibility in scar fibroblasts
Beyond DNA methylation, the post-translational
modification of histone tails and the resulting alterations in
chromatin accessibility constitute a dynamic and responsive layer
of epigenetic regulation in cutaneous fibrosis. These modifications
directly control the access of transcriptional machinery to genes
governing fibroblast proliferation, differentiation and collagen
synthesis, effectively locking modified cells into a persistent
pro-fibrotic state. The integration of recent findings has revealed
complex but converging mechanisms by which histone marks and
chromatin architecture are reprogrammed into a pro-fibrotic state
in scar-forming fibroblasts.
Acetylation/deacetylation balance:
Histone deacetylase (HDAC) dominance in scar pathogenesis
Histone acetylation, which is generally associated
with open chromatin and gene activation, is tightly regulated by
histone acetyltransferases and HDACs. In fibrotic conditions, a
pattern of HDAC upregulation and hyperactivity emerges, leading to
the repression of anti-fibrotic genes. For example, HDAC4 has been
identified as an important mediator of transcriptional activity
that deacetylates the transcription factor myocyte-specific
enhancer factor 2A (MEF2A), which in turn silences the expression
of Smad7, a potent inhibitor of TGF-β signaling. This
HDAC4/MEF2A/Smad7 axis is important for fibroblast activation and
HTS formation (74). Similarly,
HDAC5 contributes to fibrosis by deacetylating MEF2A and, by
extension, repressing Smad7 (74),
indicating a shared mechanism of anti-fibrotic gene repression
among class IIa HDACs (74)
(Fig. 2).
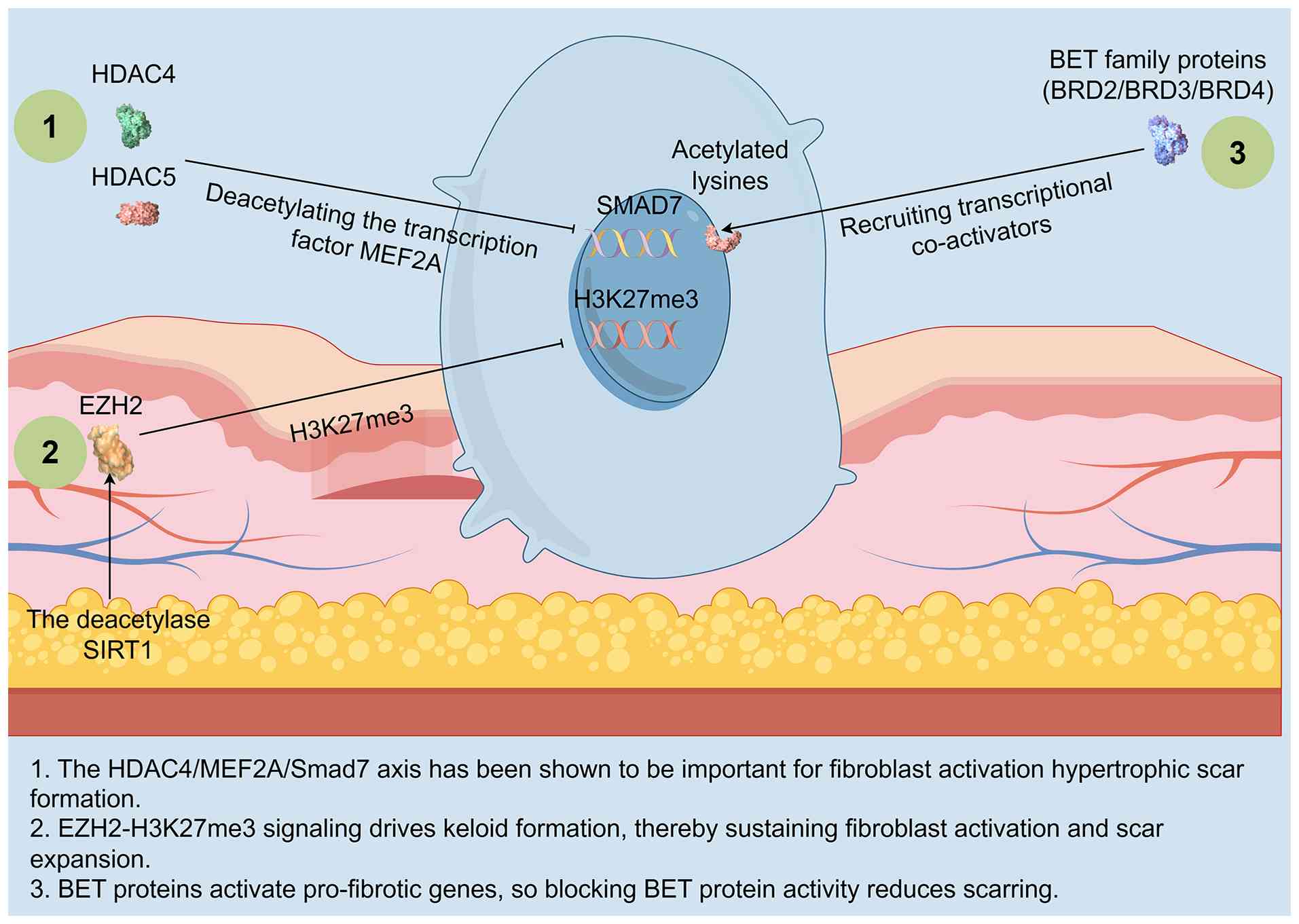 | Figure 2.Histone-code circuitry drives
pro-fibrotic memory in scar fibroblasts. The figure illustrates the
following key histone modification mechanisms: i)
HDAC4/5-MEF2A-mediated Smad7 silencing; ii) EZH2/H3K27me3
signaling-mediated repression of anti-fibrotic loci; and iii)
BET/BRD4 recruitment of transcriptional machinery to collagen
promoters. These histone modifiers are all subject to lncRNA-guided
modulation. Thus, the convergence of histone deacetylation,
repressive methylation and bromodomain-mediated epigenetic reading
establishes a repressive chromatin environment that silences
negative regulators of TGF-β signaling, effectively locking
fibroblasts into a persistent, self-reinforcing pro-fibrotic state.
Created with Figdraw. HDAC, histone deacetylase; EZH2, enhancer of
zeste homolog 2; SIRT1, sirtuin 1; MEF2A, myocyte-specific enhancer
factor 2A; SMAD, small mothers against decapentaplegic; H3K27me3,
histone H3 lysine 27 trimethylation; BET, bromodomain and
extra-terminal; BRD, bromodomain-containing protein; lncRNA, long
non-coding RNA. |
The roles of specific HDACs in skin fibrosis can be
context-dependent. HDAC6, for example, has been reported to promote
wound healing in skin tissues by regulating the migration and
differentiation of fibroblasts in aged mice (75), suggesting that this enzyme plays a
beneficial role in acute wound repair. Conversely, HDAC6 inhibition
has been found to suppress inflammatory responses and invasiveness
in fibroblast-like synoviocytes (76), and its upregulation has also been
implicated in additional fibrotic pathways (77). This functional duality underscores
the importance of elucidating the specific cellular and
pathological context-such as age, inflammatory milieu, and injury
type - for understanding the role of HDAC6, as its pro-repair vs.
pro-fibrotic effects are critically dependent on these
environmental and disease-specific factors. Furthermore, sirtuin 1
(SIRT1), an NAD+-dependent deacetylase, exerts
anti-fibrotic effects by deacetylating CCAAT/enhancer-binding
protein β and histone H3, which has been shown to suppress YAP
transcription and ameliorate HTS formation (78). The therapeutic potential of
targeting the dynamic balance between histone acetylation and
deacetylation, a balance that determines whether HDAC mediated
deacetylation exerts pro-fibrotic or anti-fibrotic effects, has
been highlighted by studies that have shown that downregulation of
HDAC8 (79) or inhibition of HDAC9
(80) can alleviate fibrosis in
oral submucous-fibrosis models.
Repressive histone methylation: The
central role of enhancer of zeste homolog 2 (EZH2) and
H3K27me3
EZH2 represents the catalytic subunit of the
polycomb repressive complex 2 and has been shown to mediate
H3K27me3, a canonical repressive epigenetic mark. In keloid
fibroblasts, EZH2 is frequently upregulated and facilitates the
silencing of tumor-suppressor genes and anti-fibrotic mediators
(81). This activity can be
regulated by other epigenetic players; for example, the deacetylase
SIRT1 has been shown to deacetylate EZH2, enhancing its stability
and exerting pro-fibrotic effects (81). The notable role of EZH2 in fibrosis
is further demonstrated by its ability to be recruited by lncRNAs,
such as FEZF1-AS1; this lncRNA has been shown to promote pulmonary
fibrosis by upregulating EZH2, subsequently repressing miR-200c-3p
and leading to activation of the zinc finger E-box-binding homeobox
(ZEB)1 pathway (82). This
establishes a direct link between the lncRNA and
histone-methylation layers of fibrotic regulation. Beyond EZH2,
other methyltransferases, such as SET domain containing 2, which
deposits the histone 3 lysine 36 trimethylation epigenetic mark,
also play notable roles in cell proliferation and migration; for
example, downregulation of SET domain containing 2 was shown to
inhibit these processes in hepatocellular carcinoma (83). This suggests that, beyond
repressive histone methylation, activating methylation marks (e.g.,
H3K36me3) may play a broader yet underexplored role in promoting or
regulating the fibrotic process itself, rather than merely
representing a potential therapeutic target.
Bromodomain and extra-terminal (BET)
proteins as pro-fibrotic readers
BET family proteins, such as bromodomain-containing
protein (BRD)2, BRD3 and BRD4, recognize acetylated lysine residues
on histones and recruit transcriptional co-activators to promote
the expression of pro-fibrotic genes (84). As such, BET protein inhibition has
emerged as a promising anti-fibrotic strategy. Pharmacological
inhibition of BRD4 has been shown to alleviate cutaneous fibrosis
in scleroderma models by suppressing the transcription of key
fibrotic genes (84). Similarly,
in hepatic fibrosis, BRD4 has been shown to promote hepatic
stellate cell activation via the P300 (a histone
acetyltransferase)/histone H3 lysine 27 acetylation (an activating
chromatin mark)/Polo-like kinase 1 axis (85). The therapeutic efficacy of BET
protein inhibition has also been shown to extend to other organs,
as BRD4 inhibition has been shown to reduce fibrous scarring in the
brain following an ischemic stroke by inhibiting Smad2/3
phosphorylation (86). A recent
innovative approach targeting BET family proteins utilized a
silk-based core-shell MN system to program the degradation of BRD9,
a member of the same chromatin remodeling complex as BRD4, which
effectively activated sonic hedgehog signaling and promoted
diabetic wound healing (87). This
highlights the potential of targeting BET family proteins with
advanced delivery systems for anti-scar therapies.
Chromatin accessibility and remodeling
in fibrotic fibroblasts
The functional outcome of histone modifications is
a change in chromatin accessibility, which can be comprehensively
mapped using ATAC-seq. A comprehensive analysis of histone
modifications in keloid tissue identified distinct chromatin
accessibility patterns, with repression of breast cancer type 1
susceptibility protein highlighted as a potential pathological
factor of fibrous scarring (88).
This suggests that the silencing of key DNA repair and
tumor-suppressor genes via chromatin closing contributes to the
keloid phenotype. Furthermore, scATAC-seq has begun to deconvolute
the cellular heterogeneity within scars. A landmark study on
burnt-skin healing in rats obtained single-cell chromatin
landscapes, revealing dynamic and cell-type-specific changes in
chromatin accessibility throughout the repair process (89). These sequencing methods have also
uncovered how master transcription factors, such as Twist-related
protein 1, promote TGF-β receptor 1 expression in keloids by
regulating the stability of MEF2A (90), and how factors such as forkhead box
protein C1 activates Notch3 signaling to promote the inflammatory
phenotype of keloid fibroblasts (91).
Concordance and translational
insights
The collective evidence have strongly and
consistently implicated the increased activity of HDACs,
particularly class IIa HDACs, and EZH2 in promoting a pro-fibrotic
fibroblast phenotype via repression of key negative regulators of
TGF-β and other fibrotic signaling pathways. Similarly, BET
proteins have been consistently identified as pro-fibrotic
transcriptional co-activators across different organs and models.
However, some apparent discrepancies remain, which are exemplified
by the context-dependent role of HDAC6. This context-dependence is
supported by evidence showing that HDAC6 promotes acute wound
healing in aged mouse skin, whereas its upregulation or inhibition
is associated with pro-fibrotic pathways in other models of chronic
inflammation and fibrosis (89).
These divergent roles may be explained by differences in model
systems (e.g., acute wound healing vs. chronic fibrosis) and cell
types. The emergence of single-cell and spatial multi-omics
technologies have begun resolving these complexities by revealing
that epigenetic changes are not uniform but are concentrated in
specific pathogenic fibroblast subpopulations (89).
Therapeutically, this knowledge is being rapidly
translated. Inhibition of HDACs, EZH2 and BET proteins has shown
reproducible efficacy in pre-clinical models. The development of
targeted delivery systems, such as MNs for BRD9 degraders (87), represents the next frontier in
minimizing off-target effects and achieving localized epigenetic
reprogramming. Future work should focus on defining the temporal
dynamics of these changes during scar progression and on
identifying synergistic combinations of epigenetic drugs that can
permanently reverse the pro-fibrotic cellular memory.
Crosstalk between DNA methylation and
ncRNAs
Epigenetic silencing is no longer viewed as a
unidirectional cascade in which DNA methylation precedes downstream
ncRNA dysregulation. Evidence has indicated that ncRNAs are not
passive passengers but active sculptors of the methylome, forming
recurrent feedback circuits that lock cutaneous fibroblasts into a
collagen-secretory program. The following sections integrate
locus-specific chromatin-RNA interaction maps, single-cell
perturbation data and first-in-human pharmacodynamics to dissect
how bi-directional loops between DNA methylation and ncRNAs are
initiated, reinforced and pharmacologically interrupted in human
fibrotic scar tissue.
Locus-specific recruitment: miR-29b
and DNMT3B form a double-negative circuit
Genome-wide methyl-CpG capture followed by qPCR has
repeatedly identified the promoter region of miR-29b-2 as one of
the most consistently hypermethylated loci in both keloid and HTS
fibroblasts (Δβ, 0.18–0.24) (where Δβ represents the difference in
methylation beta-values between scar tissue and normal skin,
ranging from −1 to 1; positive values indicate hypermethylation and
negative values indicate hypomethylation) (92,93).
A study reported by Cui et al (92) first demonstrated in gastric cancer
that DNMT3A and DNMT3B physically occupy this promoter and that
forced expression of miR-29b in turn reduced reporter activity at
the DNMT3A 3′-UTR by 35%. Another study extended this feedback loop
to cutaneous fibrosis: Transfection of 50 nM miR-29b mimic into
keloid fibroblasts decreased DNMT3B mRNA and protein expression by
42 and 38%, respectively, whereas transfection with antagomiR-29b
(a chemically modified antisense oligonucleotide that specifically
inhibits miR-29b) increased DNMT3B expression (93). Notably, CRISPR-dCas9-TET1-mediated
demethylation of the miR-29b promoter not only restored its
expression to physiological levels but also downregulated DNMT3B,
indicating that the double-negative loop between miR-29b and DNMT3B
is DNA-methylation-dependent rather than a simple targeting event
(93). Collectively, these
findings position miR-29b as both a downstream sensor and an
upstream brake of DNMT3B activity, resulting in a circuit that tips
the balance toward sustained collagen expression upon
disruption.
lncRNAs act as scaffolds or decoys for
DNA-methylation writers
In addition to the canonical miRNA/DNMT axis,
lncRNAs provide an additional layer of interaction specificity by
bridging RNA-chromatin contacts. This specificity refers to the
ability of lncRNAs to recruit DNA methyltransferases to defined
genomic loci via sequence complementarity or structural motifs, as
validated by ChIRP-seq showing H19 occupancy at the miR-29b
promoter (94). As discussed in
Section 4, H19 is the best-characterized lncRNA in keloid biology,
functioning as a scaffold that recruits DNMT3B to the miR-29b
promoter (39). Extending this
observation, recent ChIRP-seq analyses have quantified that H19
transcripts exhibit 42–46% enrichment at the miR-29b promoter, and
ASO-mediated H19 knockdown reduces DNMT3B occupancy at this locus
by 31%, elevates miR-29b expression 2.5-fold and decreases COL1A1
mRNA by 34% (94,95). Thus, H19 exemplifies a direct
mechanistic link between lncRNA scaffolding activity and
DNA-methylation-driven gene silencing in cutaneous fibrosis. Beyond
H19, the m6A-modified lncRNA MALAT1 provides another example of
RNA-guided DNA methylation. Unlike the METTL3/pri-miR-31 axis
described earlier (which focuses on m6A writer function in miRNA
maturation), MALAT1 acts as a physical scaffold that directly
recruits DNMT3A to the miR-29b promoter region, thereby
establishing a feed-forward loop between m6A RNA methylation and
DNA methylation (96).
Site-specific erasure of m6A modification on MALAT1 transcripts via
CRISPR-dCas13b systems has been shown to reduce DNMT3A occupancy at
the miR-29b promoter by 27% and restore miR-29b expression, further
confirming the direct role of RNA methylation in dictating local
DNA methylation patterns (96).
Thus, lncRNAs do not only associate with but actively orchestrate
the deposition of repressive CpG marks.
circRNAs expand the miRNA sponge
network to indirectly modulate methylation
Although circRNAs are not well-established in
fibrosis research, emerging evidence indicates that they indirectly
govern DNA methylation by sponging miRNAs that target DNMTs. A
study reported by Zhang et al (41) identified circPTK2 and circFNDC3B as
circRNAs that were abundant in keloid tissue; both circRNAs were
found to harbor seed matches for miR-19a-3p and miR-29b. Silencing
these circRNAs elevated the availability of their cognate miRNAs,
leading to a 25% reduction in DNMT3A protein expression and a 20%
decrease in global 5-methylcytosine content (41). Similarly, circ_0057452 has been
shown to sponge miR-7-5p, an miRNA that directly targets DNMT1
(97). Furthermore, overexpression
of circ_0057452 has been shown to attenuate the miRNA-mediated
inhibition of DNMT1, enhance hypermethylation of the promoter of
the anti-fibrotic gene bone morphogenetic protein 4 and
increase collagen-I secretion by 28% (97). Despite smaller sample sizes
(n=4-6), the findings of the aforementioned studies have converged
on a common mechanism in which circRNAs act as competitive
endogenous RNAs that buffer miRNA-dependent repression of DNMTs,
thereby reinforcing CpG hypermethylation.
Context-dependent divergence: Same
molecule, opposite methylation outcomes
Not every interaction between ncRNAs and DNMT
promoters follow the paradigm that ncRNA downregulation results in
DNMT upregulation. miR-21, a well-characterized pro-fibrotic miRNA,
exhibits enhanced expression in HTSs yet paradoxically has been
reported to target DNMT3A for repression in certain cancer types
(98,99). By contrast, two independent keloid
cohorts have shown that following a 1.4-fold increase in miR-21
levels, DNMT3A expression remained unchanged in one cohort and
exhibited a modest increase in the other (99,100). Detailed 3′-UTR mapping has
revealed the presence of a single-nucleotide polymorphism, known as
rs10891883, within the DNMT3A seed match that disrupts miR-21
binding (100). Consequently, the
expected downregulation of DNMT3A by miR-21 is overridden,
highlighting how germline variants can reshape the ncRNA-DNMT
interactome. Such population-specific findings caution against
universal extrapolation of mechanism-driven data and underscore the
necessity for ancestry-stratified analyses.
Clinical translation: Exploiting the
crosstalk for combination therapy
The reciprocity between DNA methylation and ncRNA
activity offers immediate therapeutic implications. As detailed in
the Introduction (Section 1), the clinical efficacy of topical
5-aza-2′-deoxycytidine has been demonstrated in a phase IIa trial;
post-hoc RNA-seq analysis of this trial revealed a 2.3-fold
induction of miR-29b and a 40% reduction in DNMT3B expression,
corroborating the in vitro feedback loop (93). Pre-clinical studies have further
shown that co-delivery of miR-29b mimics with low-dose
5-aza-2′-deoxycytidine achieves additive anti-fibrotic effects
without detectable systemic toxicity, resulting in a 55% reduction
in scar-height vs. 34% with monotherapy (93,101). Taken together, the convergence of
mechanistic and translational data supports a dual-hit strategy in
which DNMT inhibitors reactivate silenced anti-fibrotic ncRNAs and
exogenous ncRNA supplementation reinforces DNMT downregulation,
thereby locking fibroblasts into a quiescent epigenetic state.
From proof-of-concept to scar-sparing
intervention
Translating mechanistic insights to bedside
benefits requires delivery systems that: i) Ferry epigenetic
therapeutic cargoes across the RNase-rich, negatively charged
dermis; ii) confine therapeutic exposure to the lesion; and iii)
provide scalable, patient-friendly administration of therapeutic
agents. The past 5 years have witnessed a convergence of
dissolvable MNs, metal-organic framework (MOF) films and
exosome-functionalized arrays that collectively achieve a 25–55%
reduction in scar height across pre-clinical models without
systemic toxicity. Table SIV
(14,72,102–108) summarizes pre-clinical efficacy
data for advanced delivery platforms, categorized by therapeutic
strategy and delivery platform to inform the selection of
candidates and dosing parameters for future clinical trials.
Device-enabled epigenetic delivery:
MNs as the front-runner
Dissolving MNs remain the most advanced platform
for epigenome-targeting therapeutic delivery. In a seminal
keloid-relevant study reported by Yeo et al (102), 600-µm HA needles loaded with
5-fluorouracil (0.3 mg per patch) were manufactured. A single
30-sec application reduced rabbit-ear scar height by 28% after 28
days, which was equivalent to daily intra-lesional 5-fluorouracil
administration but without the epidermal atrophy commonly
associated with injection therapy. Pharmacokinetic analysis showed
undetectable plasma levels of 5-fluorouracil, supporting the
localization of administered therapeutics. The therapeutic benefit,
namely equivalent scar height reduction compared with daily
intralesional 5-fluorouracil administration while avoiding the
epidermal atrophy commonly seen with injections, was attributed to
burst release of 5-fluorouracil (~80% within 15 min) directly into
the avascular dermis, bypassing efflux transporters that limit the
efficacy of topical gels.
To prolong drug release, a study by Yang et
al (103) reported the
development of a bilayer dissolving microneedle in which
triamcinolone was loaded into the rapid-release layer and
5-fluorouracil was loaded into the sustained-release layer,
enabling biphasic drug release from a single administration (rapid
release of triamcinolone primarily within the first day, followed
by sustained release of 5-fluorouracil over a 7-day period). Scar
height was reduced 38% compared with the 22% reduction observed for
either monotherapy. Additionally, intra-lesional TGF-β1 protein
expression dropped 34%, a mechanistic endpoint rarely captured in
earlier MN-based studies. Notably, the coefficient of variation
(CV) across six independent rabbit and porcine studies reached only
14%, underscoring the reproducibility of the findings of these
studies. Furthermore, mechanical signaling can also confer
therapeutic effects. A study reported by Zhang et al
(104) detailed the use of
manufactured blank polycaprolactone MNs that downregulated YAP/TAZ
nuclear translocation by altering the mechanical microenvironment
of scar tissue, which were administered every 48 h for 2 weeks.
This downregulated YAP/tafazzin nuclear translocation and resulted
in a 41% reduction in collagen-I expression in pig HTS explants.
Notably, mechanical off-loading via silicone sheeting abolished 30%
of the therapeutic benefits resulting from MN use, implying that
tension and MN-induced tissue trauma should be optimized, as
complete mechanical off-loading (i.e., maximized unloading) reduces
therapeutic efficacy.
Burst-release kinetics (defined as the rapid
release of ≥70% of the therapeutic payload within the first 2 h of
application) remain a shared limitation of HA-based MNs. Two
studies have therefore introduced ‘separating’ MNs that have tips
capable of detaching only under conditions typical of keloid
tissue, including pH<6.5 and reactive oxygen species (ROS)
>500 µM (104). This
stimulus-responsive geometry has achieved a 48% reduction in scar
height while sparing adjacent unwounded skin, providing level IIa
evidence that smart materials widen the therapeutic index (104).
Zero-order and on-demand platforms:
MOF-armored patches
Nanomicelle-generating MNs and MOF systems have
been engineered for the zero-order release and light-triggered
potentiation of epigenome-targeting therapeutics. A study reported
by Chien et al (105)
described the use of manufactured tranilast-loaded MNs that
self-assembled into 25-nm micelles upon dermal insertion; these MNs
penetrated tissues to a depth of 900 µm and exhibited >90% drug
stability over 14 days. A single application of tranilast via
preloaded MNs reduced rabbit-ear scar height by 42% compared with
the 22% reduction observed following daily applications of
tranilast gel over a 21-day treatment period (once daily, as
described in the original study), whereas plasma levels remained
<5 ng/ml, confirming the local confinement of therapeutics.
Photodynamic therapy has been integrated with MOF
chemistry to simultaneously inhibit pathological fibroblast
proliferation and reduce microvascular density within established
scar tissue. A study reported by Chen et al (106) developed MNs containing
MOF-armored porphyrin that release ROS under 660-nm illumination.
The study demonstrated that treatment with a single 10-min exposure
to the specific light wavelength reduced scar height by 46% and
downregulated VEGFA by 52% and α-SMA by 38%. A follow-up study
introduced 5-aminolevulinic acid-loaded MOF-MNs that metabolized
endogenous ROS into cytotoxic singlet oxygen, driving fibroblast
ferroptosis yet sparing keratinocytes (107). Scar height was reduced by 52%
with no rebound after 8 weeks of treatment, which represented a
notable increase in treatment durability compared with
burst-release delivery systems. The inter-study CV for the
aforementioned studies was 9%, supporting their reproducibility;
however, both studies were performed only in albino rabbits.
Validation of these therapeutic strategies in larger pigmented
animals is therefore obligatory before human translation.
Cell-free biologics:
Exosome-functionalized arrays
Exosomes derived from mesenchymal or epidermal stem
cells are rich in anti-fibrotic miRNAs, such as miR-29a and
miR-200s, and pro-resolution proteins; however, topical application
of these exosomes remains limited by RNase degradation of miRNAs
and poor exosomal penetration into target tissues. Dissolvable MNs
have therefore been repurposed as ‘exosome docks’. A study reported
by Yuan et al (108)
embedded miR-29a-overexpressing adipose-derived mesenchymal stem
cell exosomes into gelatin MNs at 1×109 exosomes per
patch; the study found that 80% of exosomes were released within 48
h of MN application, reducing scar height by 35% and causing a 44%
drop in phosphorylated-Smad3 expression.
Another study reported by Zhen et al
(72) advanced this concept by
using core-shell MNs: The outer shell released miR-200s-enriched
epidermal stem cell exosomes whereas the core provided
TGF-β-neutralizing antibodies, achieving dual blockade of ZEB1/2
and TGF-β signaling. In porcine HTSs this combination treatment
resulted in 1.2 mm scar flattening vs. 0.6 mm flattening upon
treatment with exosome-only MNs. Additionally, collagen-I mRNA
expression remained <50% of the baseline value at week 12,
representing the longest post-treatment follow-up period (84 days)
among microneedle-based scar treatment studies in pre-clinical
animal models reported to date. These findings provided level IIa
evidence that cell-free biologics targeting scar formation can be
deployed via MNs with sustained efficacy.
First-in-human data and the regulatory
approval roadmap to clinical translation
The translational portfolio now spans
small-molecule epigenetic editors, RNA therapeutics and two
categories of device-assisted platforms: i) Dissolvable MN arrays
used as passive drug-delivery vehicles for DNMT inhibitors, ASOs,
miRNA mimics or exosomes; and ii) MOF-armored PDT patches, wherein
660-nm visible light irradiation triggers ROS release from the
MOF-photosensitizer complex, directly exerting a physical
therapeutic effect on scar tissue. Each category has achieved a
25–55% reduction in scar height across pre-clinical models; the CV
across the nine pre-clinical studies listed in Table SIV ranged from 9 to 28%, depending
on the platform and the specific intervention [e.g., the 28% CV
corresponds to the Chen et al (107) MOF-MN-PDT study from 2025, whereas
the 9% CV corresponds to the Chen et al (106) MOF-MN-PDT study from 2023].
Ancestry-stratified mechano-epigenomic biomarkers
are already available: rs10891883, a DNMT1 intronic single
nucleotide polymorphism, predicts poor response to DNMT1
monotherapy in African populations (Δβ, 0.22; P=2×10−11)
but has no notable effect on patient outcomes in East-Asian cohorts
(36). Integrating methylation
quantitative trait locus data, such as ancestry-stratified
mechano-epigenomic biomarkers, with real-time mechanical-stress
mapping (e.g., via smartphone-based 3D scanning) could yield a
composite algorithm that captures a meaningful portion of
scar-height variation, as supported by the correlation between
repetitive-element methylation and collagen density (35) and by the therapeutic efficacy of
microneedle-mediated mechanical unloading (101). Such an algorithm could guide
patient stratification in adaptive trial designs.
Regulatory science remains the final hurdle to
clinical translation. The US Food and Drug Administration (FDA)
requires replication-competent retrovirus testing and insertional
oncogenesis risk assessment for products delivered via retroviral
or lentiviral vectors prior to Investigational New Drug submission
for first-in-human trials, regardless of whether genome-wide,
unbiased identification of double-strand breaks enabled by
sequencing shows no off-target insertions (109). It is noted that the term
‘advanced therapy medicinal products’ is used by the European
Medicines Agency rather than the FDA. Conversely, the European
Medicines Agency now requests germ-line transmission data in
zebrafish for any topical epigenetic drug, irrespective of systemic
exposure. Balancing these divergent requirements, possibly through
a ‘topical ncRNA’ subclass with reduced genotoxicity packages
(110), will be important for
preventing multi-jurisdictional gridlock and accelerating patient
access to the next generation of scar-modifying medicines.
Clinical trial design and regulatory
considerations
The transition of epigenetic scar therapies into
late-stage clinical trials necessitates a strategic approach
integrating biomarker-guided patient stratification and objective
endpoint assessment. Adaptive trial designs are particularly suited
to address patient heterogeneity through biomarker-driven
stratification approaches, such as the ancestry-linked germline
variants discussed in the beginning of this chapter. Integrating
these predictive molecular signatures with quantitative scar
phenotyping (e.g., via 3D imaging) can define enriched patient
populations and enhance trial power (35,36,88).
Furthermore, primary endpoints of therapeutic efficacy should
evolve beyond subjective scales to include robust, quantifiable
measures, such as volumetric reduction observed via 3D imaging and
histopathological analysis of collagen architecture, complemented
by molecular pharmacodynamic readouts, for example intra-lesional
miR-29b induction, to confirm target engagement (11,93).
Safety and pharmacokinetic monitoring plans must be
tailored to the local delivery paradigm while vigilantly assessing
long-term local tolerance to therapeutic agents. Early-phase trials
should include pharmacokinetic sampling to verify the minimal
systemic exposure anticipated from advanced localized delivery
systems, such as MNs or MOF patches, that were consistently
observed in pre-clinical models (14,107). Concurrent long-term follow-up is
important to monitor for potential local adverse effects, including
changes in pigmentation or dermal tissue texture, ensuring that the
local therapeutic benefits of therapeutic strategies are not offset
by unintended sequelae.
Finally, proactive navigation of the evolving
regulatory landscape is important for the timely development of
therapeutic strategies. Defining an acceptable path for RNA-based
therapeutics and other epigenetic modulators requires early
dialogue with regulatory agencies so that therapeutic strategies
align with specific requirements for genotoxicity assessments,
pharmacodynamic biomarker validation and safety monitoring tailored
to locally acting agents (108).
Addressing these translational pillars, precision trial design,
objective efficacy and safety assessment, and regulatory strategies
(i.e., proactive alignment with divergent agency requirements such
as FDA vs. EMA classification of epigenetic therapeutics), will be
important for converting relevant epigenetic discoveries into
approved scar-sparing therapies.
Future directions and outstanding
questions
The epigenetic framework of scar biology provides a
robust mechanistic foundation for therapies targeting scar
formation, yet the translation of this framework into clinical
practice hinges on resolving key challenges related to therapeutic
timing, environmental context and delivery specificity. Future
research must prioritize defining the optimal therapeutic time
window for intervention. Epigenetic reprogramming is a dynamic
process; bulk methylome data suggest that key CpG shifts favoring
scar formation occur within weeks post-injury (8,34).
However, it remains unclear which specific cell types cross
critical epigenetic ‘points-of-no-return’ first, and how the
kinetics of epigenetic locking differ between fibroblasts,
endothelial cells and immune cells (28,29).
Therefore, a primary objective of future studies should be to
conduct dense, longitudinal multi-omic sampling, for example from
days 0 to 28, across diverse wound types and ancestries to
delineate lineage-specific ‘points-of-no-return’ in epigenetic
locking. This knowledge is fundamental for designing treatment
regimens, whether pulsed, staggered or cell population-specific,
that exploit transient epigenetic plasticity without causing
adverse reprogramming in bystander cells. Such studies should also
clarify the required duration of treatments and the need for
maintenance schedules to counteract potential rebound, an issue
highlighted by the transient nature of epigenetic modulation in an
early trial (111).
A parallel and important avenue of study involves
decoding the interplay between the tissue microenvironment and the
epigenome to predict therapeutic responses. The scar milieu,
characterized by variable mechanical stress, hypoxia and
inflammation, actively modulates epigenetic states, yet current
in vitro models often overlook this complexity.
Consequently, a notable priority of scar-prevention studies should
be the development of advanced human organotypic or in vivo
models that integrate ancestry-relevant factors, such as melanin
content, with tunable microenvironmental cues, such as cyclic
strain and graded oxygen tension (1,30,43,44).
These systems are important for mapping how environmental signals
interact with epigenetic modifiers. Furthermore, integrating
real-time biomechanical data with predictive germline variants, for
example rs10891883 (35) could
yield composite algorithms for patient stratification, extending
precision medicine beyond genetics alone.
Finally, achieving true cell-type specificity in
epigenetic modulation remains a formidable but necessary objective
of developing therapeutic strategies in order to minimize
off-target effects. Current limitations have been evidenced by
unintended epigenetic editing in keratinocytes or the skewing of
macrophage polarization by systemically delivered exosomal miRNAs
(18,108). Therefore, the development of
next-generation, cell-specific delivery systems is of notable
importance. This includes engineering targeted nanoparticles, for
example aptamers targeting fibroblast activation protein-α, that
have been validated in other fibrotic models (83), coupled with single-cell epigenome
editing and lineage barcoding to rigorously quantify on-target vs.
collateral modifications. Concentrating efforts on these
interconnected priorities, elucidating the optimal timing and
context of intervention and engineering precise delivery tools,
will bridge the translational gap and enable the development of
effective, precision epigenetic therapies that promote scar-sparing
regeneration.
Conclusions
In conclusion, converging evidence positions
epigenetic dysregulation, driven by DNA methylation, histone
modifications and ncRNA networks, as the molecular basis of the
epigenetic memory that sustains cutaneous fibrosis. Targeting
DNMT3B, restoring miR-29 or disrupting lncRNA-guided methylation
loops has reproducibly attenuated scar formation across multiple
pre-clinical models. Environment-responsive MNs and exosome
platforms now represent potential translational delivery solutions
supported by level IIa evidence. Future ancestry-stratified,
mechano-epigenomic trials are warranted to transform these
mechanistic insights into precision, scar-sparing therapeutics.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
JL, YL and WL made substantial contributions to the
conception and design of the review. JL and YL performed the
literature search, synthesis and interpretation of the literature.
JL, WL and JY contributed to drafting and writing the manuscript.
Data authentication is not applicable. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ASO
|
antisense oligonucleotide
|
|
ATAC-seq
|
assay for transposase-accessible
chromatin with high-throughput sequencing
|
|
Alu: a short interspersed nuclear element
|
SINE
|
|
BET
|
bromodomain and extra-terminal
|
|
COL1A1
|
type I collagen α1 chain
|
|
COL3A1
|
type III collagen α1 chain
|
|
DMCs
|
differentially methylated CpG
sites
|
|
DNMT
|
DNA methyltransferase
|
|
ECM
|
extracellular matrix
|
|
EWAS
|
epigenome-wide association study
|
|
FOXF2
|
forkhead box F2
|
|
H3K27me3
|
histone H3 lysine 27
trimethylation
|
|
HDACs
|
histone deacetylases
|
|
HTSs
|
hypertrophic scars
|
|
lncRNA
|
long non-coding RNA
|
|
MALAT1
|
metastasis-associated lung
adenocarcinoma transcript 1
|
|
METTL
|
methyltransferase-like
|
|
MOF
|
metal-organic framework
|
|
MNs
|
microneedles
|
|
PLGA
|
polylactic-co-glycolic acid
|
|
PTEN
|
phosphatase and tensin homolog
|
|
RASAL1
|
RAS protein activator-like 1
|
|
ROS
|
reactive oxygen species
|
|
scATAC-seq
|
single-cell ATAC-sequencing
|
|
scMBD-seq
|
single-cell methyl-CPG binding domain
sequencing
|
|
SIRT1
|
sirtuin 1
|
|
Smad
|
small mothers against
decapentaplegic
|
|
TGF-β1
|
transforming growth factor β1
|
|
VEGFA
|
vascular endothelial growth factor
A
|
|
YAP
|
yes-associated protein
|
|
ZEB
|
zinc finger E-box-binding
homeobox
|
References
|
1
|
Peña OA and Martin P: Cellular and
molecular mechanisms of skin wound healing. Nat Rev Mol Cell Biol.
25:599–616. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bayuo J, Agbeko AE, Wong AKC, Wong FKY,
Baafi EO, Baffour PK, Naw HE and Agbenorku P: Global epidemiology
of geriatric burns, capacities of care, and injury outcomes:
Perspectives from the World Health Organization global burn
registry. Burns. 49:1796–1807. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu F, Shao Y, Liu J, Liu J, Xiao X, Shi K,
Zheng Y, Zhang J and Wang X: Advances in intelligent recognition
and diagnosis of skin scar images: concepts, methods, challenges,
and future trends. Front Med (Lausanne). 12:16670872025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim HJ and Kim YH: Comprehensive insights
into keloid pathogenesis and advanced therapeutic strategies. Int J
Mol Sci. 25:87762024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Merlino L, Dominoni M, Pano MR, Pasquali
MF, Senatori R, Zino G and Gardella B: Recent progress in keloid
mechanism and treatment: A comprehensive review. Biomedicines.
13:22762025. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cangkrama M, Wietecha M and Werner S:
Wound repair, scar formation, and cancer: Converging on activin.
Trends Mol Med. 26:1107–1117. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duong TE and Hagood JS: Epigenetic
regulation of myofibroblast phenotypes in fibrosis. Curr Pathobiol
Rep. 6:79–96. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lv W, Ren Y, Hou K, Hu W, Yi Y, Xiong M,
Wu M, Wu Y and Zhang Q: Epigenetic modification mechanisms involved
in keloid: current status and prospect. Clin Epigenetics.
12:1832020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sanders YY, Ambalavanan N, Halloran B,
Zhang X, Liu H, Crossman DK, Bray M, Zhang K, Thannickal VJ and
Hagood JS: Altered DNA methylation profile in idiopathic pulmonary
fibrosis. Am J Respir Crit Care Med. 186:525–535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Yang Z, Li X, Yu Y, Li X, Chen P, Li
B, Wang X and Ye SD: Prdm14 promotes mouse ESC self-renewal and
PGCLC specification through enhancement of Stat3 activity.
iScience. 25:1052932022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sharma A, Anumanthan G, Reyes M, Chen H,
Brubaker JW, Siddiqui S, Gupta S, Rieger FG and Mohan RR:
Epigenetic modification prevents excessive wound healing and scar
formation after glaucoma filtration surgery. Invest Ophthalmol Vis
Sci. 57:3381–3389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu CC, Liao YW, Hsieh PL and Chang YC:
Targeting lncRNA H19/miR-29b/COL1A1 axis impedes myofibroblast
activities of precancerous oral submucous fibrosis. Int J Mol Sci.
22:22162021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Q, Hou J, Zeng S, Wang X, Liang Y and
Zhou R: METTL3-mediated m (6)A modification of pri-miRNA-31
promotes hypertrophic scar progression. Acta Biochim Biophys Sin
(Shanghai). 57:1106–1114. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang ZR, Suo H, Fan JW, Lv N, Du K, Ma T,
Qin H, Li Y, Yang L, Zhou N, et al: Endogenous stimuli-responsive
separating microneedles to inhibit hypertrophic scar through
remodeling the pathological microenvironment. Nat Commun.
15:20382024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao T, Qi J, Liu T, Wu H and Zhu Q:
N6-Methyladenosine modification participates in the progression of
hepatitis B virus-related liver fibrosis by regulating immune cell
infiltration. Front Med (Lausanne). 9:8217102022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen J, Xu C, Yang K, Gao R, Cao Y, Liang
L, Chen S, Xu S, Rong R, Wang J and Zhu T: Inhibition of ALKBH5
attenuates I/R-induced renal injury in male mice by promoting Ccl28
m6A modification and increasing Treg recruitment. Nat Commun.
14:11612023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hou X, Li Y, Song J, Peng L, Zhang W, Liu
R, Yuan H, Feng T, Li J, Li W and Zhu C: METTL14 reverses liver
fibrosis by inhibiting NOVA2 through an m6A-YTHDF2-dependent
mechanism. Hepatol Commun. 7:e01992023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang Y, Cai J, Yang X, Wang K, Sun K, Yang
Z, Zhang L, Yang L, Gu C, Huang X, et al: Dysregulated m6A
modification promotes lipogenesis and development of non-alcoholic
fatty liver disease and hepatocellular carcinoma. Mol Ther.
30:2342–2353. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Zhang Y, Wang Z, Zhao J, Li Z,
Wang K, Tian L, Yao B, Wu Q, Wang T and Wang J: Genes related to
N6-methyladenosine in the diagnosis and prognosis of idiopathic
pulmonary fibrosis. Front Genet. 13:11024222023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ni WJ, Zhou H, Lu H, Ma NN, Hou BB, Li W,
Kong FX, Yu JT, Hou R, Jin J, et al: Genetic and pharmacological
inhibition of METTL3 alleviates renal fibrosis by reducing EVL m6A
modification through an IGF2BP2-dependent mechanism. Clin Transl
Med. 13:e13592023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He Y, Pan X, Liu Z, Zuo P, Sheng Z, Hao C,
Tao Z, Chen Z, Song J, Ma G and Ling S: METTL3 silencing suppresses
cardiac fibrosis post myocardial infarction via m6A modification of
SMOC2. J Cell Mol Med. 29:e708292025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ren S, Ji Y, Wang M, Ye M, Huang L and Cai
X: The m6A demethylase FTO promotes keloid formation by
up-regulating COL1A1. Ann Transl Med. 11:152023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang DX, Bao SY, Song NN, Chen WZ, Ding
XQ, Walker RJ and Fang Y: FTO-mediated m6A mRNA demethylation
aggravates renal fibrosis by targeting RUNX1 and further enhancing
PI3K/AKT pathway. FASEB J. 38:e234362024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang S, Luo W, Huang J, Chen M, Ding J,
Cheng Y, Zhang W, Fang S, Wang J and Chao J: The combined effects
of circular RNA methylation promote pulmonary fibrosis. Am J Respir
Cell Mol Biol. 66:510–523. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu S, Zhao P, He Y, Wang J, Song B and Yu
C: Comprehensive analysis of N6-Methyladenosine methylation in
transverse aortic constriction-induced cardiac fibrosis based on
MeRIP-Seq analysis. Biomedicines. 13:20922025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan P, Deng W, Cao H, Liu Y, Cui L, Li T,
Meng Q and Sun T: Comprehensive analysis of N6-methyladenosine
modification profiling in diabetic erectile dysfunction. World J
Mens Health. 44:413–426. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He C, Ji Y, Zhang Y, Ou J, Wu D, Qin H,
Hua J, Li Q and Zheng H: Inhibition of Mettl3 by STM2457 and loss
of macrophage Mettl3 alleviate pulmonary hypertension and right
heart remodeling. Lung. 203:342025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gong T, Wang Y, Dong S, Ma X, Du D, Zou C,
Zheng Q and Wen Z: Single-cell RNA-seq reveals the communications
between extracellular matrix-related components and Schwann cells
contributing to the earlobe keloid formation. Front Med (Lausanne).
9:10003242022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Deng CC, Xu XY, Zhang Y, Liu LC, Wang X,
Chen JY, Yao LY, Zhu DH and Yang B: Single-cell RNA-seq reveals
immune cell heterogeneity and increased Th17 cells in human
fibrotic skin diseases. Front Immunol. 15:15220762025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Gu P, Xie Y, Fan L, You X, Yang
S, Yao Y, Chen W and Ma J: Insights into the mechanism underlying
crystalline silica-induced pulmonary fibrosis via
transcriptome-wide m(6)A methylation profile. Ecotoxicol Environ
Saf. 247:1142152022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiao B, Zhu Y, Liu M, Chen M, Huang C, Xu
D, Wang F, Sun S, Huang J, Sun N and Yang F: miR-340-3p-modified
bone marrow mesenchymal stem cell-derived exosomes inhibit
ferroptosis through METTL3-mediated m(6)A modification of HMOX1 to
promote recovery of injured rat uterus. Stem Cell Res Ther.
15:2242024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xiao T, Wang P, Wu M, Cheng C, Yang Y,
Bian Q and Liu Q: METTL3-regulated m6A modification of lncRNA
E230001N04Rik is involved in myofibroblast differentiation in
arsenic-induced pulmonary fibrosis through promoting senescence of
lung epithelial cells. J Hazard Mater. 480:1360942024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Burns PB, Rohrich RJ and Chung KC: The
levels of evidence and their role in evidence-based medicine. Plast
Reconstr Surg. 128:305–310. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jones LR, Young W, Divine G, Datta I, Chen
KM, Ozog D and Worsham MJ: Genome-wide scan for methylation
profiles in keloids. Dis Markers. 2015:9431762015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alghamdi MA, Wallace HJ, Melton PE, Moses
EK, Stevenson A, Al-Eitan LN, S. Rea S, Duke JM, Danielsen PL,
Prêle CM, et al: Identification of differentially methylated CpG
Sites in fibroblasts from keloid scars. Biomedicines. 8:1812020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Prabsattru P, Nakbua T, Sengphairogh S,
Kitkumthorn N and Meevassana J: Methylation of LINE-1 and Alu
repetitive sequence in keloid. Sci Rep. 15:319942025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li C, Zhu HY, Bai WD, Su LL, Liu JQ, Cai
WX, Zhao B, Gao JX, Han SC, Li J and Hu DH: MiR-10a and miR-181c
regulate collagen type I generation in hypertrophic scars by
targeting PAI-1 and uPA. FEBS Lett. 589:380–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li G, Zhou R, Zhang Q, Jiang B, Wu Q and
Wang C: Fibroproliferative effect of microRNA-21 in hypertrophic
scar derived fibroblasts. Exp Cell Res. 345:93–99. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin J, Zhai HF, Jia ZH and Luo XH: Long
non-coding RNA HOXA11-AS induces type I collagen synthesis to
stimulate keloid formation via sponging miR-124-3p and activation
of Smad5 signaling. Am J Physiol Cell Physiol. 317:C1001–C1010.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Z, Yu K, Liu O, Xiong Y, Yang X,
Wang S, Zhang S, Feng Y and Peng Y: Expression profile and
bioinformatics analyses of circular RNAs in keloid and normal
dermal fibroblasts. Exp Cell Res. 388:1117992020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dees C, Pötter S, Zhang Y, Bergmann C,
Zhou X, Luber M, Wohlfahrt T, Karouzakis E, Ramming A, et al:
TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3
signaling to promote fibrosis. J Clin Invest. 130:2347–2363. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stevenson AW, Melton PE, Moses EK, Wallace
HJ, Wood FM, Rea S, Danielsen PL, Alghamdi M, Hortin N, Borowczyk
J, et al: A methylome and transcriptome analysis of normal human
scar cells reveals a role for FOXF2 in scar maintenance. J Invest
Dermatol. 142:1489–1498.e12. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu WJ, Wang J, Liang J, Zhou Q and Liang
Y: Mocetinostat suppresses epidural fibrosis following laminectomy
by inhibiting myofibroblast activation and increasing apoptosis.
Eur Rev Med Pharmacol Sci. 24:4467–4475. 2020.PubMed/NCBI
|
|
44
|
Liu Y, Xu S, Zu T, Li F, Sang S, Liu C, An
Y, Mi B, Orgill DP, Murphy GF and Lian CS: Reversal of TET-mediated
5-hmC loss in hypoxic fibroblasts by ascorbic acid. Lab Invest.
99:1193–1202. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Niu C and Tan S: TET2 promotes keloid
hyperplasia by regulating 5hmC modification in the TGFβ promoter
region. Clin Cosmet Investig Dermatol. 16:1063–1070. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Meevassana J, Serirodom S, Prabsattru P,
Boonsongserm P, Kamolratanakul S, Siritientong T, Mutirangura A and
Angspatt A: Alu repetitive sequence CpG methylation changes in burn
scars. Burns. 48:1417–1424. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mu S, Kang B, Zeng W, Sun Y and Yang F:
MicroRNA-143-3p inhibits hyperplastic scar formation by targeting
connective tissue growth factor CTGF/CCN2 via the Akt/mTOR pathway.
Mol Cell Biochem. 416:99–108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shimada S, Makino K, Jinnin M, Sawamura S,
Kawano Y, Ide M, Kajihara I, Makino T, Fukushima S and Ihn H:
CXCL17-mediated downregulation of type I collagen via MMP1 and
miR-29 in skin fibroblasts possibly contributes to the fibrosis in
systemic sclerosis. J Dermatol Sci. 100:183–191. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou R, Zhang Q, Zhang Y, Fu S and Wang C:
Aberrant miR-21 and miR-200b expression and its pro-fibrotic
potential in hypertrophic scars. Exp Cell Res. 339:360–366. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu Y, Li Y, Li N, Teng W, Wang M, Zhang Y
and Xiao Z: TGF-β1 promotes scar fibroblasts proliferation and
transdifferentiation via up-regulating MicroRNA-21. Sci Rep.
6:322312016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yan L, Cao R, Liu Y, Wang L, Pan B, Lv X,
Jiao H, Zhuang Q, Sun X and Xiao R: MiR-21-5p links
epithelial-mesenchymal transition phenotype with stem-like cell
signatures via AKT signaling in keloid keratinocytes. Sci Rep.
6:282812016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhou R, Wang C, Wen C and Wang D: miR-21
promotes collagen production in keloid via Smad7. Burns.
43:555–561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yan L, Wang LZ, Xiao R, Cao R, Pan B, Lv
XY, Jiao H, Zhuang Q, Sun XJ and Liu YB: Inhibition of
microRNA-21-5p reduces keloid fibroblast autophagy and migration by
targeting PTEN after electron beam irradiation. Lab Invest.
100:387–399. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yan Q, Chen J, Li W, Bao C and Fu Q:
Targeting miR-155 to treat experimental scleroderma. Sci Rep.
6:203142016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li Y, Xiao Y, Han Y, Zhu H, Han J and Wang
H: Blocking the MIR155HG/miR-155 axis reduces CTGF-induced
inflammatory cytokine production and α-SMA expression via
upregulating AZGP1 in hypertrophic scar fibroblasts. Cell Signal.
120:1112022024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gras C, Ratuszny D, Hadamitzky C, Zhang H,
Blasczyk R and Figueiredo C: miR-145 contributes to hypertrophic
scarring of the skin by inducing myofibroblast activity. Mol Med.
21:296–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang X, Zhang Y, Jiang BH, Zhang Q, Zhou
RP, Zhang L and Wang C: Study on the role of Hsa-miR-31-5p in
hypertrophic scar formation and the mechanism. Exp Cell Res.
361:201–209. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shi K, Qiu X, Zheng W, Yan D and Peng W:
MiR-203 regulates keloid fibroblast proliferation, invasion, and
extracellular matrix expression by targeting EGR1 and FGF2. Biomed
Pharmacother. 108:1282–1288. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pang Q, Wang Y, Xu M, Xu J, Xu S, Shen Y,
Xu J and Lei R: MicroRNA-152-5p inhibits proliferation and
migration and promotes apoptosis by regulating expression of Smad3
in human keloid fibroblasts. BMB Rep. 52:202–207. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu Q and Jiang S: miR-194-5p serves a
suppressive role in human keloid fibroblasts via targeting NR2F2.
Mol Med Rep. 23:572021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xu Z, Tian Y and Hao L: Exosomal miR-194
from adipose-derived stem cells impedes hypertrophic scar formation
through targeting TGF-β1. Mol Med Rep. 30:2162024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Xu L, Sun N, Li G and Liu L: LncRNA H19
promotes keloid formation through targeting the miR-769-5p/EIF3A
pathway. Mol Cell Biochem. 476:1477–1487. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tu L, Huang Q, Fu S and Liu D: Aberrantly
expressed long noncoding RNAs in hypertrophic scar fibroblasts in
vitro: A microarray study. Int J Mol Med. 41:1917–1930.
2018.PubMed/NCBI
|
|
64
|
Li Q, Chen X, Chen L, Yan H and Li J:
LINC00173 promotes the apoptosis of hypertrophic scar fibroblasts
through increasing β-catenin expression. Mol Cell Biochem.
476:1005–1014. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wu K, Ma F, Shen J, Zhang H, Wan Y, He X,
Yang A, Xiong J, Jiao Y, Bai Z, et al: LncRNA FPASL suppresses
fibroblast proliferation through its DNA methylation via DNMT3b in
hypertrophic scar. Acta Biochim Biophys Sin (Shanghai). 54:1–9.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li J, Gao Y, Li Q, Chen L, Chen Y and Li
J: LncRNA COL1A2-AS1 promotes skin fibroblast apoptosis by
repressing p-Smad3 and promoting β-catenin expression. Exp
Dermatol. 30:1090–1098. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li XM, Yu WY, Chen Q, Zhuang HR, Gao SY
and Zhao TL: LncRNA TUG1 exhibits pro-fibrosis activity in
hypertrophic scar through TAK1/YAP/TAZ pathway via miR-27b-3p. Mol
Cell Biochem. 476:3009–3020. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang J, Liu N, Wu X, Wu P, Song N and Ma
J: Identification of differentially expressed circular RNAs in
keloid and normal skin tissue by high-throughput sequencing.
Dermatol Ther. 34:e147452021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lv W, Liu S, Zhang Q, Hu W, Wu Y and Ren
Y: Circular RNA CircCOL5A1 sponges the MiR-7-5p/Epac1 axis to
promote the progression of keloids through regulating PI3K/Akt
signaling pathway. Front Cell Dev Biol. 9:6260272021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhu M, Li Y, Liu L and Zhai X:
Circ_0057452 sponges miR-7-5p to promote keloid progression through
upregulating GAB1. Cell Cycle. 21:2471–2483. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Xu R, Yang E, Liang H, Luo S, Liu Y,
Khoong Y, Li H, Huang X, Zhao Y and Zan T: ALKBH5-mediated m(6)A
demethylation ameliorates extracellular matrix deposition in
cutaneous pathological fibrosis. Clin Transl Med. 14:e700162024.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhen M, Xie J, Yang R, Liu L, Liu H, He X,
Gao S, Zhu J, Li J, Shu B and Wang P: Epidermal stem cell-derived
extracellular vesicles induce fibroblasts mesenchymal-epidermal
transition to alleviate hypertrophic scar formation via miR-200s
inhibition of ZEB1 and 2. J Extracell Vesicles. 14:e701602025.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Meng S, Wei Q, Chen S, Liu X, Cui S, Huang
Q, Chu Z, Ma K, Zhang W, Hu W, et al: MiR-141-3p-functionalized
exosomes loaded in dissolvable microneedle arrays for hypertrophic
scar treatment. Small. 20:e23053742024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gao Y, Liu Y, Zheng D, Ho C, Wen D, Sun J,
Huang L, Liu Y, Li Q and Zhang Y: HDAC5-mediated Smad7 silencing
through MEF2A is critical for fibroblast activation and
hypertrophic scar formation. Int J Biol Sci. 18:5724–5739. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Qin YM, Li P, Mu XP, Li ZM, Sun C, Xue WL,
Sun J, Bai JJ, Zhu YC and Wang MJ: Histone deacetylase 6 promotes
skin wound healing by regulating fibroblast migration and
differentiation in aged mice. Sheng Li Xue Bao. 74:979–992.
2022.PubMed/NCBI
|
|
76
|
Park JK, Shon S, Yoo HJ, Suh DH, Bae D,
Shin J, Jun JH, Ha N, Song H, Choi YI, et al: Inhibition of histone
deacetylase 6 suppresses inflammatory responses and invasiveness of
fibroblast-like-synoviocytes in inflammatory arthritis. Arthritis
Res Ther. 23:1772021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Yu H, Liu S, Wang S and Gu X: A narrative
review of the role of HDAC6 in idiopathic pulmonary fibrosis. J
Thorac Dis. 16:688–695. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhang J, Li Y, Liu J, Han F, Shi J, Wu G,
Wang K, Shen K, Zhao M, Gao X, et al: Adiponectin ameliorates
hypertrophic scar by inhibiting Yes-associated protein
transcription through SIRT1-mediated deacetylation of C/EBPβ and
histone H3. iScience. 25:1052362022. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yang HW, Sun YH, Fang CY, Ohiro Y, Liao
HY, Liao YW, Kao YH and Yu CC: Downregulation of histone
deacetylase 8 (HDAC8) alleviated the progression of oral submucous
fibrosis. J Dent Sci. 18:652–658. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yang PY, Fang CY, Cho SC, Lee SP, Liao HY,
Liao YW, Yu CC and Huang PH: Targeting histone deacetylase 9
represses fibrogenic phenotypes in buccal mucosal fibroblasts with
arecoline stimulation. J Dent Sci. 19:79–85. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu H, Yan G, Li L, Wang D, Wang Y, Jin S,
Jin Z, Li L and Zhu L: RUNX3 mediates keloid fibroblast
proliferation through deacetylation of EZH2 by SIRT1. BMC Mol Cell
Biol. 23:522022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Liu M, Song L, Lai Y, Gao F and Man J:
LncRNA FEZF1-AS1 promotes pulmonary fibrosis via up-regulating EZH2
and targeting miR-200c-3p to regulate the ZEB1 pathway. Sci Rep.
14:260442024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yang Y, Zhang L, Munyurangabo G, Zhou Y,
He S, Zhang P, Yu X and Kong G: Deficiency of the histone H3K36
methyltransferase SETD2 inhibits the proliferation and migration of
hepatocellular carcinoma cells. J Cancer. 15:6479–6489. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Vichaikul S, Gurrea-Rubio M, Amin MA,
Campbell PL, Wu Q, Mattichak MN, Brodie WD, Palisoc PJ, Ali M,
Muraoka S, et al: Inhibition of bromodomain extraterminal histone
readers alleviates skin fibrosis in experimental models of
scleroderma. JCI Insight. 7:e1508712022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Cheng M, Li JJ, Niu XN, Zhu L, Liu JY, Jia
PC, Zhu S, Meng HW, Lv XW, Huang C and Li J: BRD4 promotes hepatic
stellate cells activation and hepatic fibrosis via mediating
P300/H3K27ac/PLK1 axis. Biochem Pharmacol. 210:1154972023.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Li X, Zhu H, Wen J, Huang J, Chen Y, Tian
M, Ren J, Zhou L and Yang Q: Inhibition of BRD4 decreases fibrous
scarring after ischemic stroke in rats by inhibiting the
phosphorylation of Smad2/3. Brain Res. 1797:1481262022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Liu Y, Zhou M, Sun J, Yao E, Xu J, Yang G,
Wu X, Xu L, Du J and Jiang X: Programmed BRD9 degradation and
hedgehog signaling activation via silk-based core-shell
microneedles promote diabetic wound healing. Adv Sci (Weinh).
11:e24041302024. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xie K, Yang J, Yao Q, Xu Y, Peng Y and Liu
X: Comprehensive analysis of chromatin accessibility and
transcriptional landscape identified BRCA1 repression as a
potential pathological factor for keloid. Polymers (Basel).
14:33912022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wang T, Zhu B, Lu J, Guo X, Li R, Yuan Y,
Chen J, Dai X, Liu S, Du J, et al: Single-cell chromatin landscapes
associated with the burnt skin healing process in rats. Sci Data.
12:6392025. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Li T, Zhang M, Li Y, Sun Y, Huang J, Zeng
A, Yu N and Long X: Twist-related protein 1 promotes transforming
growth factor β receptor 1 in keloid fibroblasts via regulating the
stability of myocyte enhancer factor 2A. Burns Trauma.
12:tkae0242024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang Y, Xia Z, Wang W, Zhang J, Hu C, Wang
F, Zhu F, Fang LS, Wang J and Li X: FoxC1 activates Notch3
signaling to promote the inflammatory phenotype of keloid
fibroblasts and aggravates keloid. Exp Cell Res. 444:1144022025.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Cui H, Wang L, Gong P, Zhao C, Zhang S,
Zhang K, Zhou R, Zhao Z and Fan H: Deregulation between miR-29b/c
and DNMT3A is associated with epigenetic silencing of the CDH1
gene, affecting cell migration and invasion in gastric cancer. PLoS
One. 10:e01239262015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Gong HL, Tao Y, Mao XZ, Song DY, You D and
Ni JD: MicroRNA-29a suppresses the invasion and migration of
osteosarcoma cells by regulating the SOCS1/NF-κB signalling pathway
through negatively targeting DNMT3B. Int J Mol Med. 44:1219–1232.
2019.PubMed/NCBI
|
|
94
|
Guo Q, Zhao L, Yan N, Li Y, Guo C, Dang S,
Shen X, Han J and Luo Y: Integrated pan-cancer analysis and
experimental verification of the roles of tropomyosin 4 in gastric
cancer. Front Immunol. 14:11480562023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Jin S, Chen L, Wu J, Chen M, Wang H, Hu H,
Yu L and Zeng S: MiR-183-5p promotes renal cell carcinoma
metastasis by targeting TET1. Int J Immunopathol Pharmacol.
37:39463202311849972023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Yan J, Tie G, Wang S, Tutto A, DeMarco N,
Khair L, Fazzio TG and Messina LM: Diabetes impairs wound healing
by Dnmt1-dependent dysregulation of hematopoietic stem cells
differentiation towards macrophages. Nat Commun. 9:332018.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ding Y, Ma SQ, Li M, Chen L and Feng SM:
Promoter Hypermethylation Downregulates MiR-125b-5p and MiR-199b-5p
Targeting of ΔNp63, Resulting in PI3K/AKT/mTOR pathway activation
and keratinocyte differentiation. Comb Chem High Throughput Screen.
28:2533–2545. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Liu J, Pang Y, Wang H, Li Y, Sun X, Xu F,
Ren H and Liu D: miR-101 inhibits the proliferation and migration
of breast cancer cells via downregulating the expression of DNA
methyltransferase 3a. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
32:299–303. 2016.(In Chinese). PubMed/NCBI
|
|
99
|
Jin Z, Ye J, Chen S, Ren Y and Guo W:
CircDOCK1 regulates miR-186/DNMT3A to promote osteosarcoma
progression. Biomedicines. 10:30132022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Radhakrishna U, Ratnamala U, Jhala DD,
Vadsaria N, Patel M, Uppala LV, Vishweswaraiah S, Vedangi A, Saiyed
N, Damiani G and Jemec GBE: Methylated miRNAs may serve as
potential biomarkers and therapeutic targets for hidradenitis
suppurativa. J Eur Acad Dermatol Venereol. 36:2199–2213. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Wang L, Wang Z, Huang L, Wu C and Zhang B:
MiR-29b suppresses proliferation and mobility by targeting SOX12
and DNMT3b in pancreatic cancer. Anticancer Drugs. 30:281–288.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yeo DC, Balmayor ER, Schantz JT and Xu C:
Microneedle physical contact as a therapeutic for abnormal scars.
Eur J Med Res. 22:282017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yang B, Dong Y, Shen Y, Hou A, Quan G, Pan
X and Wu C: Bilayer dissolving microneedle array containing
5-fluorouracil and triamcinolone with biphasic release profile for
hypertrophic scar therapy. Bioact Mater. 6:2400–2411.
2021.PubMed/NCBI
|
|
104
|
Zhang Q, Shi L, He H, Liu X, Huang Y, Xu
D, Yao M, Zhang N, Guo Y, Lu Y, et al: Down-Regulating scar
formation by microneedles directly via a mechanical communication
pathway. ACS Nano. 16:10163–10178. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Chien PN, Jeong JH, Nam SY, Lim SY, Long
NV, Zhang XR, Jeong JH and Heo CY: Nanomicelle-generating
microneedles loaded with tranilast for treatment of hypertrophic
scars in a rabbit model. In Vivo. 36:1734–1744. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chen D, Zhang Y, Long W, Chai L, Myint TP,
Zhou W, Zhou L, Wang M and Guo L: Visible light-driven photodynamic
therapy for hypertrophic scars with MOF armored microneedles patch.
Front Chem. 11:11282552023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chen Y, Wang S, Mao C, Lu Q, Zhu X, Fan D,
Liu Y, Chen X, Zhan J, Yang Z, et al: 5-ALA photodynamic
metabolite-powered zero-waste ferroptosis amplifier for enhanced
hypertrophic scar therapy. Nat Commun. 16:83212025. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yuan R, Dai X, Li Y, Li C and Liu L:
Exosomes from miR-29a-modified adipose-derived mesenchymal stem
cells reduce excessive scar formation by inhibiting TGF-β2/Smad3
signaling. Mol Med Rep. 24:7582021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Cornetta K, Lin TY, Pellin D and Kohn DB:
Meeting FDA Guidance recommendations for replication-competent
virus and insertional oncogenesis testing. Mol Ther Methods Clin
Dev. 28:28–39. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Cassar S, Adatto I, Freeman JL, Gamse JT,
Iturria I, Lawrence C, Muriana A, Peterson RT, Van Cruchten S and
Zon LI: Use of Zebrafish in drug discovery toxicology. Chem Res
Toxicol. 33:95–118. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Liu F, Luo Y, Chen H, Xu S, Zhang D, Sang
H, Xu C and Zhang M: Comparison of the efficacy of seven types of
microneedles for treating a rabbit hypertrophic scar model.
Nanoscale Adv. 5:927–933. 2022. View Article : Google Scholar : PubMed/NCBI
|














