Introduction
Non-small cell lung cancer (NSCLC) is a significant
cause of cancer-related mortality in the US and worldwide (1). Epidermal growth factor receptor (EGFR)
is overexpressed in various epithelial types of cancer, including
NSCLC, and its inhibitors have been investigated as first-line or
subsequent therapy options for patients with advanced metastatic
NSCLC (2). Two classes of EGFR
inhibitor, monoclonal antibodies, including cetuximab, and
small-molecule tyrosine kinase inhibitors (TKIs), including
erlotinib and gefitinib, have been studied in phase III trials and
are used clinically to treat NSCLC (3–8).
Although erlotinib is an effective therapy for NSCLC, resistance to
erlotinib reduces its efficacy. Erlotinib resistance is associated
with changes in EGFR itself or in the expression of other genes
(9). In particular, the T790M
mutation has been reported in 50% of EGFR TKI-resistant tumors
(10,11). To investigate the basis of erlotinib
resistance, erlotinib-resistant human NSCLC A549 cells, termed
A549/ER cells, were isolated. A PCR array was performed to identify
erlotinib resistance in A549/ER cells.
Materials and methods
Reagents
Paclitaxel, ethyl methanesulfonate and 3-[4,
5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT) were
obtained from Sigma Chemical Co. (St. Louis, MO, USA). The
gemcitabine hydrochloride used was from LKT Laboratories, Inc. (MN,
USA). Erlotinib was from Chemie Tek (Indianapolis, IN, USA). Fetal
bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM),
penicillin-streptomycin solution (10,000 U/ml penicillin and 10,000
μg/ml streptomycin) were from Hyclone (UT, USA). RT-2 Profiler PCR
Array Human Cancer Drug Resistance and Metabolism (PAHS-004A-2) was
from SA Biosciences (Frederick, MD, USA).
Cell culture
The A549 cell line, derived from NSCLC, was
maintained in DMEM containing 10% FBS, 100 U/ml penicillin and 100
μg/ml streptomycin at 37°C in a 5% CO2 humidified
atmosphere.
Cell proliferation by 3-[4,
5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide assay
Cell proliferation in vitro was measured
using a MTT colorimetric assay in 96-well plates. The cells
(5×103) were inoculated into each well. Following
overnight incubation (37°C in 5% CO2), anti-cancer
agents were added to the cultured and incubated for 3 days. A total
of 50 μl of MTT (1 mg/ml) was added to each well and the plates
were incubated for an additional 4 h. Following aspiration of the
culture medium, the resulting formazan was dissolved with 100 μl of
dimethylsulfoxide. The plates were read at 570 nm using a
microplate reader.
Chronic erlotinib exposure
Erlotinib-resistant A549/ER cells were isolated by
the A549 cells with increasing concentrations of erlotinib
following ethyl methanesulfonate-induced mutagenesis, and then
incubated in a selection medium with erlotinib (1–100 μM).
Reverse transcription-polymerase chain
reaction (RT-PCR) method
Total cellular RNA was extracted by the RNeasy Mini
kit (Qiagen Sciences, MD, USA). RNA quality and concentration were
confirmed in NanoDrop ND-100 Spectrophotometer (Thermo Scientific,
Wilmington, DE, USA). For RT-PCR, 1 μg of total RNA was used for
cDNA synthesis using an iScript cDNA synthesis kit (Bio-Rad, CA,
USA), according to the manufacturer’s instructions. The conditions
for the RT-PCR were: 5 min at 95°C and then 28 cycles of
amplification in PCR master mix (Promega, WI, USA) at 95°C for 30
sec, annealing at 52°C for 30 sec and extension at 72°C for 1 min.
The primers used for this analysis were: p21, forward
5′-ctcttcggcccagtggacagc-3′ and reverse
5′-agagtctccaggtccacctgg-3′; fibroblast growth factor 2 (FGF2),
forward 5′-ccttgcccgaggatggcggca-3′ and reverse
5′-ttgaccggtaagtattgtagt-3′; EGFR, forward 5′-gccacaggccaggt
ctgccat-3′ and reverse 5-ccggcgtctgcgtacttccag-3; and GAPDH,
forward 5′-gtcttcaccaccatggagaagg-3′ and reverse
5′-ggcaggtcaggtccaccactga-3′.
Genomic DNA extraction and single
nucleotide polymorphism genotyping
Genomic DNA from A549 and A549/ER cells were
extracted with a QIAamp DNA Mini kit (Qiagen Sciences). The
conditions for the RT-PCR were: 5 min at 95°C and then 55 cycles of
amplification in PCR master mix (Promega) at 95°C for 30 sec,
annealing at 52°C for 30 sec and extension at 72°C for 1 min. The
mutations were genotyped using PSQ96MA (Qiagen, Germantown, MD,
USA).
The primers used for this analysis were: T790M,
forward 5′-tgggcatctgcctcacct-3′, reverse 5′-ctttgtgttcccggacat-3′
and sequence primer 5′-cctcacctccaccgt-3′; L861Q, forward
5′-agccaggaacgtactggtgaa-3′, reverse 5′-gcctccttctgcatggtattc-3′
and sequence primer 5′-tcacagattttgggc-3′.
PCR array
Total cellular RNA was extracted by the RNeasy Mini
kit (Qiagen Sciences). RNA quality and concentration were confirmed
in the NanoDrop ND-100 Spectrophotometer (Thermo Scientific). For
RT-PCR, 1 μg of total RNA was used for cDNA synthesis using the
iScript cDNA synthesis kit (Bio-Rad), according to the
manufacturer’s instructions. After cDNAs were mixed with SYBR-Green
Supermix, the mixtures were added to the plates of RT-2 Profiler
PCR Array Human Cancer Drug Resistance and Metabolism. The
conditions for real-time PCR were: 10 min at 95°C and then 40
cycles at 95°C for 15 sec and at 60°C for 1 min. The data from PCR
array were normalized according to the manufacturer’s guidelines
using software from SA Bioscience.
Statistical analysis
Data are presented as the mean ± SD. Statistical
analysis was performed using StatView 5.0. (SAS Institute Inc.,
Cary, NC, USA). Differences were considered significant at
P<0.05.
Results
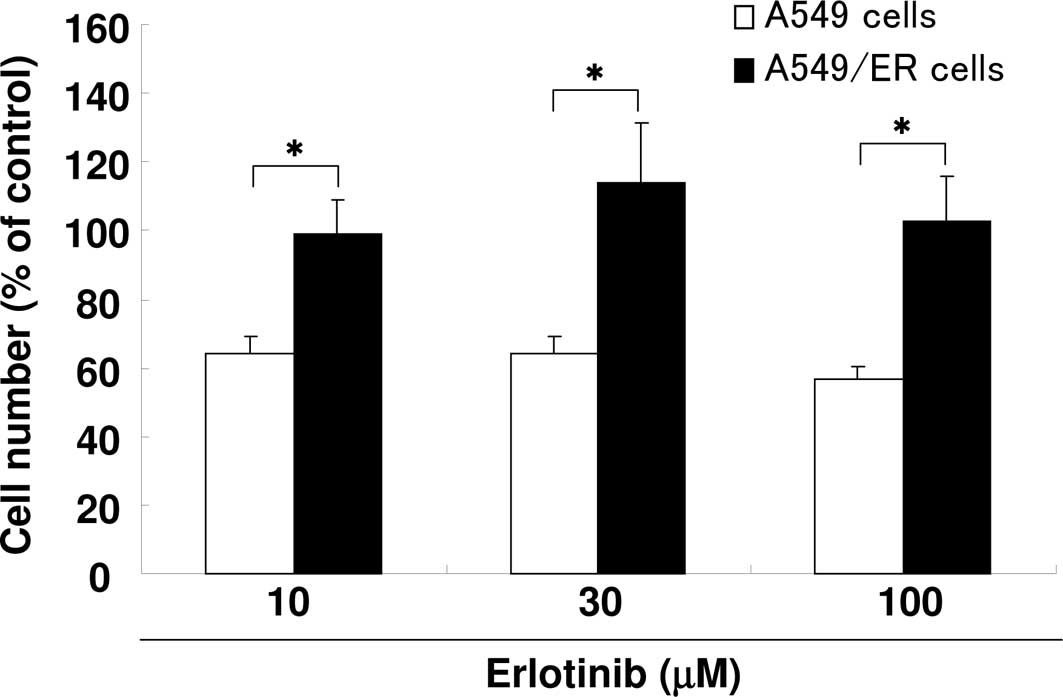
Establishment of erlotinib-resistant
non-small cell lung cancer A549 cells
To isolate erlotinib-resistant A549/ER cells, A549
cells were cultured in stepwise selection containing an increasing
erlotinib concentration, from 10 to 100 μM. The sensitivity to
erlotinib was examined in each cell line. Results showed that the
A549/ER cells were more resistant than the parental A549 cells
(Fig. 1).
EGFR mutation in A549/ER cells
EGFR mutations, including the
threonine-to-methionine substitution at position 790 (T790M, in
exon 20) and the leucine-to-glutamine (L861Q, in exon 21) have been
reported to be resistance mutations to erlotinib (12). To examine T790M and L861Q mutations
in A549/ER cells, pyrosequencing was used. The EGFR T790M
and L861Q mutations were not present in the A549/ER cells (data not
shown).
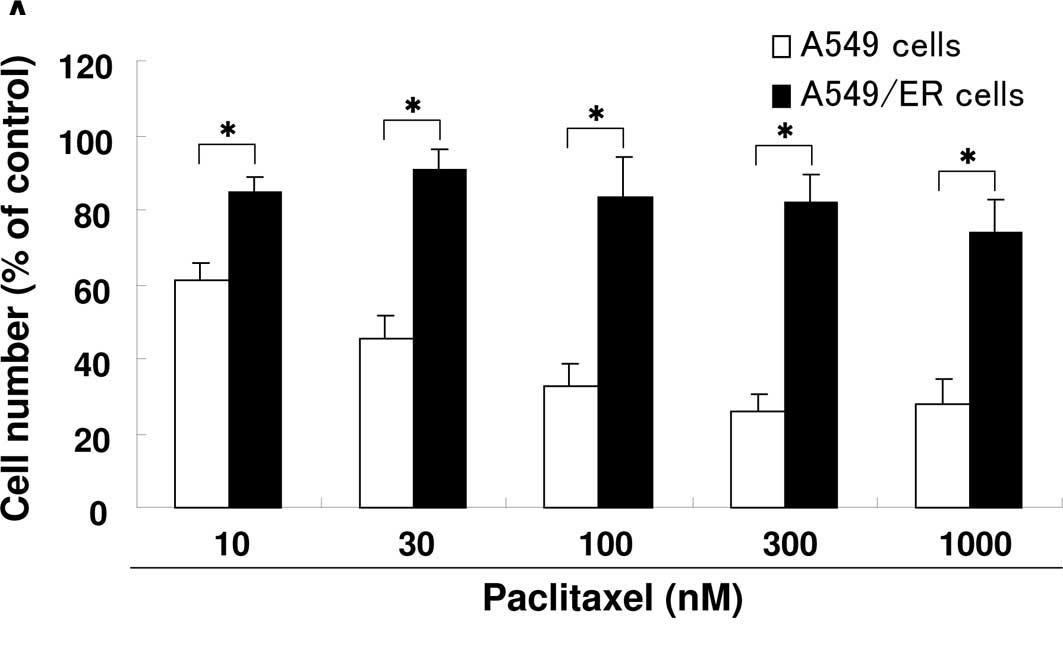
Cross resistance to erlotinib
The drug sensitivity of each cell line was tested
using a MTT assay. Fig. 2 shows the
sensitivity of the parental and resistant cell lines to various
anti-cancer agents. Notably, A549/ER cells were resistant to
paclitaxel and gemcitabine as compared to A549 cells (Fig. 2A and B).
PCR array analysis and RT-PCR analysis of
A549 and A549/ER cells
The temporal pattern of gene expression in A549 and
A549/ER cells was compared by a PCR array that covered 84 genes. A
number of genes including EGFR and MET were
down-regulated in A549/ER compared to A549 cells. A total of five
genes were up-regulated more than 1.4-fold in A549/ER cells. The
genes, including the ones for FGF2 and p21/CDKN1A,
were increased in A549/ER compared to A549 cells (Fig. 3A). The expression of insulin-like
growth factor-1 receptor (IGF1R) and IGF2R was
not altered between A549 and A549/ER cells. To validate the PCR
array using independent methods, RT-PCR analysis was performed
using specific primers for each gene. Consistent with the array
data, FGF2 and p21 expression was increased in A549
cells, while EGFR expression was decreased (Fig. 3B).
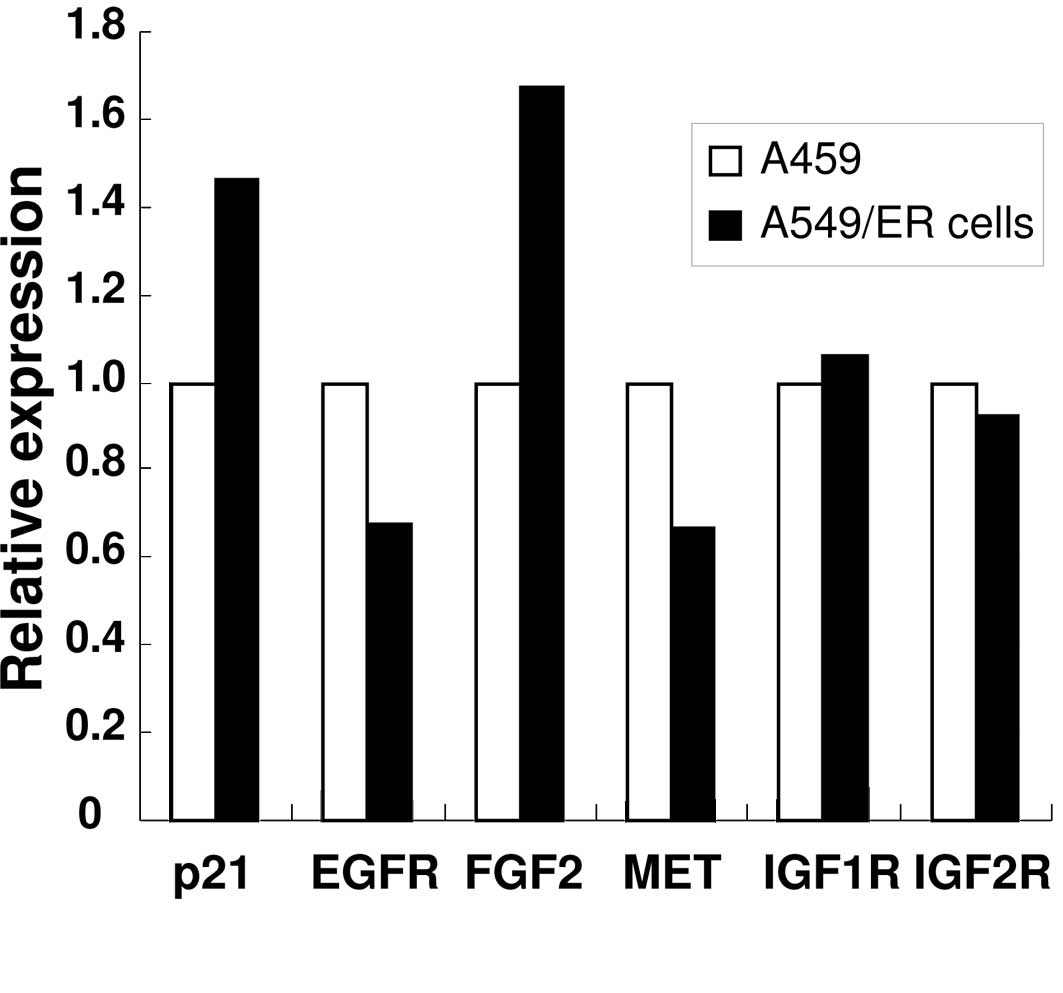 | Figure 3Expression of EGFR, MET,
p21 and FGF2 in A549 and A549/ER cells. (A)
Expression of p21, EGFR, MET, FGF2,
IGF1R and IGF2R in A549 and A549/ER cells was
detected by PCR array. Following preparation of the total RNA, 1 μg
was used for cDNA synthesis. The expression of genes was determined
by PCR array, as described in Materials and methods. Data are
normalized to the expression of each gene in parental A549 cells.
(B) Expression of EGFR, FGF2 and p21 in A549
and A549/ER cells. The expression of EGFR, FGF2,
p21 and GAPDH in A549 and A549/ER cells was measured
using RT-PCR. GAPDH expression was detected as a
control. |
Discussion
Lung cancer is the most common cause of
cancer-related mortality in developed nations. NSCLC normally
presents as incurable locally advanced or metastatic disease.
Despite significant research efforts, survival prospects remain
poor and 14% of all patients with lung cancer are expected to live
5 years after diagnosis (1).
EGFR-specific TKIs, including gefitinib and erlotinib, have been
developed as therapeutic agents for NSCLC treatment (6,7).
Although the benefits of erlotinib were statistically significant,
resistance to erloninib reduces its efficacy. To investigate the
basis of resistance to erlotinib, we isolated erlotinib-resistant
human NSCLC A549 cells, termed A549/ER cells.
One mechanism of acquired resistance occurs when
EGFR is mutated. Mutations at T790M and L861Q are normally
associated with erlotinib resistance (12). Therefore, we performed a sequence of
genome in A549 cells and A549/ER cells. Results showed that neither
A549 nor A549/ER cells carried a mutation of T790M and L861Q; thus,
the acquired resistance identified in this study was not caused by
EGFR mutation.
To investigate the basis of resistance to erlotinib,
we performed a PCR array in A549 and A549/ER cells. The expression
of EGER and MET in A549/ER cells was decreased when
compared to the sensitive A549 cells. Additionally, similar levels
of IGF1R and IGF2R were expressed in the two cell
lines. EGFR signaling is linked to multiple intracellular pathways
that inhibit apoptosis and promote survival and proliferation. Upon
ligand-induced activation, EGFR and MET generate phosphotyrosine
sites for the recruitment of Ras and phosphatidylinositol-3 kinase
(PI3K), resulting in the classic mitogen-activated protein kinase
and Akt pathway (13). These
results suggest that A549/ER cells are independent of EGFR
signaling for their growth and survival. Notably, the expression of
FGF2 in A549/ER cells was increased compared to that in A549
cells. Through the activation of FGFR1β signaling, FGF2 promotes
the survival and proliferation of tumors. Stimulation of FGFR1β
results in PI3K/Akt activation and causes resistance to anti-cancer
agents (14,15). Neutralizing FGFR1-specific antibody
abrogates the physiologic and chemoprotective effects of
FGF2/FGFR1β signaling (15). The
A549/ER cells may have alternative means of maintaining PI3K/Akt
signaling, including FGF2/FGFR overexpression, that activates
PI3K/Akt signaling in an EGFR- or MET-independent manner.
Strategies, including the neutralizing FGFR1 or FGF2-specific
antibody, or inhibitors of FGFR1, may be effective in restoring
sensitivity to erlotinib-resisitant cells.
On the other hand, the overexpression of p21
inhibits proliferation in mammalian cells and has been found to
inhibit all cyclin-CDK complexes, suggesting that it is a
cyclin-CDK inhibitor (16). p21 has
demonstrated that the inhibitory control on cyclin-CDK complexes is
mediated through its N-terminal domain and is distinct from its
ability to bind PCNA (16). p21
plays an essential role in growth arrest after DNA damage, while
its overexpression leads to cell cycle arrest, which prevents DNA
damage (17). Erlotinib-mediated
signaling is involved in the up-regulation of p21 (18). We demonstrated the overexpression of
p21 in erlotinib-resistant cells (A549/ER cells) when
compared to A549 cells (Fig. 3).
Additionally, Ferrandiz et al have reported that
p21-deficient colon cancer cells were more sensitive towards
imatinib and gefitinib than parental cells (19). These results suggest that the
overexpression of p21 may prevent erlotinib signaling in A549/ER
cells.
In conclusion, a better understanding of the
characterization and mechanism of resistance to erlotinib in
A549/ER cells may be useful in the identification of agents that
reverse clinical erlotinib resistance in NSCLC.
Acknowledgements
The authors would like to thank the University of
Wisconsin Carbone Comprehensive Cancer Center (UWCCC) for use of
its facilities to complete this study. The study is supported in
part by NIH/NCI P30 CA014520 - UW Comprehensive Cancer Center
Support.
References
|
1
|
Azzoli CG, Baker S Jr, Temin S, et al:
American Society of Clinical Oncology Clinical Practice Guideline
update on chemotherapy for stage IV non-small-cell lung cancer. J
Clin Oncol. 27:6251–6266. 2009. View Article : Google Scholar
|
|
2
|
Hirsch FR, Varella-Garcia M and Cappuzzo
F: Predictive value of EGFR and HER2 overexpression in advanced
non-small-cell lung cancer. Oncogene. 28:32–37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hynes NE and Lane HA: ERBB receptors and
cancer: the complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mendelsohn J and Baselga J: Status of
epidermal growth factor receptor antagonists in the biology and
treatment of cancer. J Clin Oncol. 21:2787–2799. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kelly K, Chansky K, Gaspar LE, Albain KS,
Jett J, Ung YC, Lau DH, Crowley JJ and Gandara DR: Phase III trial
of maintenance gefitinib or placebo after concurrent
chemoradiotherapy and docetaxel consolidation in inoperable stage
III non-small-cell lung cancer: SWOG S0023. J Clin Oncol.
26:2450–2456. 2008. View Article : Google Scholar
|
|
7
|
Gatzemeier U, Pluzanska A, Szczesna A, et
al: Phase III study of erlotinib in combination with cisplatin and
gemcitabine in advanced non-small-cell lung cancer: the Tarceva
Lung Cancer Investigation Trial. J Clin Oncol. 25:1545–1552. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pirker R, Pereira JR, Szczesna A, von
Pawel J, Krzakowski M, Ramlau R, Vynnychenko I, Park K, Yu CT,
Ganul V, Roh JK, Bajetta E, O’Byrne K, de Marinis F, Eberhardt W,
Goddemeier T, Emig M and Gatzemeier U; FLEX Study Team. Cetuximab
plus chemotherapy in patients with advanced non-small-cell lung
cancer (FLEX): an open-label randomised phase III trial. Lancet.
373:1525–1531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gazdar AF: Epidermal growth factor
receptor inhibition in lung cancer: the evolving role of
individualized therapy. Cancer Metastasis Rev. 29:37–48. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kosaka T, Yatabe Y, Endoh H, Yoshida K,
Hida T, Tsuboi M, Tada H, Kuwano H and Mitsudomi T: Analysis of
epidermal growth factor receptor gene mutation in patients with
non-small cell lung cancer and acquired resistance to gefitinib.
Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Balak MN, Gong Y, Riely GJ, Somwar R, Li
AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, Ladanyi M,
Miller VA and Pao W: Novel D761Y and common secondary T790M
mutations in epidermal growth factor receptor-mutant lung
adenocarcinomas with acquired resistance to kinase inhibitors. Clin
Cancer Res. 12:6494–6501. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kancha RK, von Bubnoff N, Peschel C and
Duyster J: Functional analysis of epidermal growth factor receptor
(EGFR) mutations and potential implications for EGFR targeted
therapy. Clin Cancer Res. 15:460–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pawson T: Specificity in signal
transduction: from phosphotyrosine-SH2 domain interactions to
complex cellular systems. Cell. 116:191–203. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song S, Wientjes MG, Gan Y and Au JL:
Fibroblast growth factors: an epigenetic mechanism of broad
spectrum resistance to anticancer drugs. Proc Natl Acad Sci USA.
97:8658–8663. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karajannis MA, Vincent L, Direnzo R,
Shmelkov SV, Zhang F, Feldman EJ, Bohlen P, Zhu Z, Sun H, Kussie P
and Rafii S: Activation of FGFR1beta signaling pathway promotes
survival, migration and resistance to chemotherapy in acute myeloid
leukemia cells. Leukemia. 20:979–986. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abukhdeir AM and Park BH: P21 and p27:
roles in carcinogenesis and drug resistance. Expert Rev Mol Med.
10:e192008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
18
|
Sutter AP, Höpfner M, Huether A, Maaser K
and Scherübl H: Targeting the epidermal growth factor receptor by
erlotinib (Tarceva) for the treatment of esophageal cancer. Int J
Cancer. 118:1814–1822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferrandiz N, Martin-Perez J, Blanco R,
Donertas D, Weber A, Eilers M, Dotto P, Delgado MD and Leon J:
HCT116 cells deficient in p21(Waf1) are hypersensitive to tyrosine
kinase inhibitors and adriamycin through a mechanism unrelated to
p21 and dependent on p53. DNA Repair. 8:390–399. 2009. View Article : Google Scholar : PubMed/NCBI
|