Introduction
Ewing sarcoma/primitive neuroectodermal tumors
(ES/PNETs) are malignant tumors whose cells typically express the
CD99 surface antigen encoded by the MIC2 gene (1). ES/PNETs most commonly arise in bone,
and occasionally originate in soft tissue (1). However, it is relatively rare for
ES/PNETs to arise from the stomach. Only four cases of gastric
ES/PNETs have been reported in the English-language literature
(PubMed, National Library of Medicine, Bethesda, MD, USA) prior to
September 2010 (2–5). In this study, the four cases were
assessed and an additional case was reported to further
characterize the clinicopathological parameters in gastric
ES/PNETs.
Case report
A 41-year-old Japanese woman was admitted with
abdominal pain in 2000 to Saiseikai Niigata Second Hospital,
Niigata, Japan. A contrast-enhanced computed tomography scan
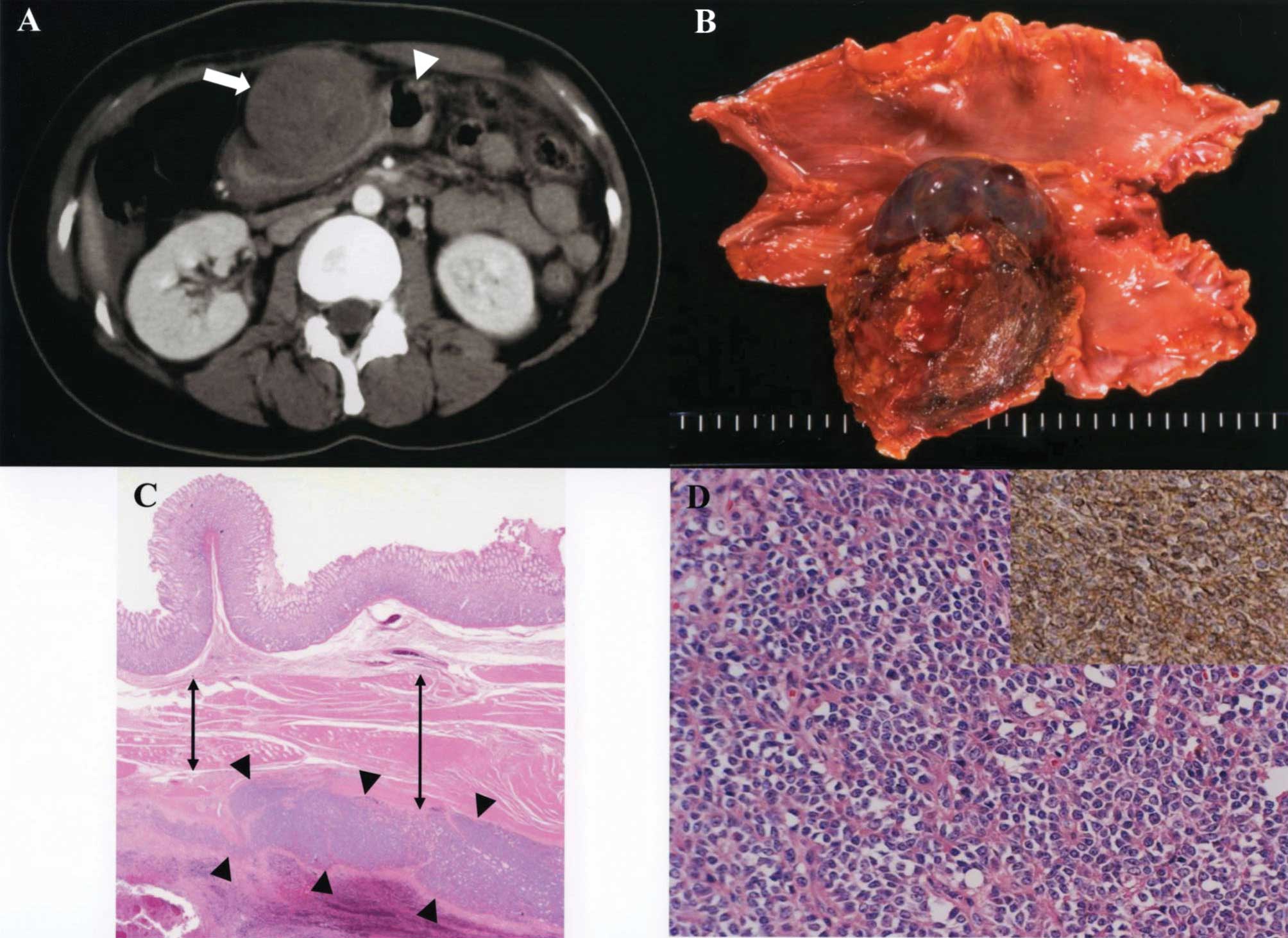
revealed a solid mass in the anterior wall of the stomach (Fig. 1A). She underwent a distal
gastrectomy and regional lymphadenectomy. The resected specimen
contained a firm, round tumor with extraluminal growth, 90×55 mm in
diameter, located in the anterior wall of the stomach (Fig. 1B). Histological examination revealed
the tumor arising from the muscularis externa of the stomach
(Fig. 1C). Tumor cells were
characterized by uniform, compact, round to oval nuclei, a modest
amount of pale eosinophilic cytoplasm, and pseudorosette formation
(Fig. 1D). Tumor cells showed
positive immunoreactivity for CD99 (Fig. 1D), vimentin, CD117 (c-kit), S100,
chromogranin A and synaptophysin. The chromosomal karyotype
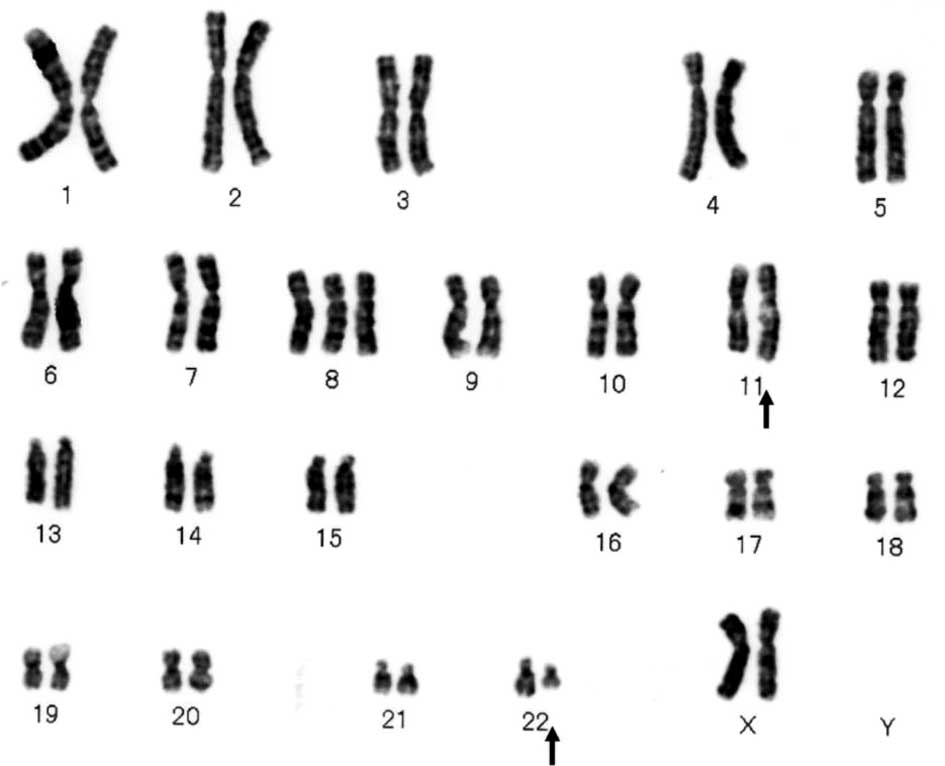
demonstrated the translocation t(11;22)(q24;q12) (Fig. 2). Molecular analysis using reverse
transcription-polymerase chain reaction revealed the
EWS-FLI1 fusion gene translocation t(11;22)(q24;q12). Thus,
the primary gastric tumor was determined to be an ES/PNET.
During the 6 years following the initial surgery,
the patient underwent five metastasectomies for isolated peritoneal
metastases. A follow-up computed tomography scan performed 6 years
post-gastrectomy showed multiple intra-abdominal peritoneal
metastases. The patient received three courses of chemotherapy
according to the modified Rosen T-16 (6) protocol, comprising ifosfamide (2000
mg/m2/day, day 1–5), etoposide (120
mg/m2/day, day 2–4), doxorubicin (30
mg/m2/day, day 5 and 6) and cyclophosphamide (1250
mg/m2/day, day 1 and 2) intravenously. A subsequent
computed tomography scan revealed that the tumor had responded
partially to the treatment as defined by the Response Evaluation
Criteria in Solid Tumors (RECIST) criteria (7). Metastasectomies were then performed to
remove multiple peritoneal metastases. Following these surgeries,
the patient received five courses of chemotherapy, which followed
the modified Rosen T-16 protocol, over a 5-month period. The
patient then underwent three metastasectomies for isolated
peritoneal metastases. In 2008, a follow-up computed tomography
scan identified para-aortic lymph node metastases. The patient
received radiation therapy for the recurrent nodal disease, and a
computed tomography scan revealed a partial response by the tumor
to this treatment as defined by RECIST criteria. However, despite
treatment, the growth of the peritoneal metastases progressed and
para-aortic and mediastinal lymph node metastases appeared. The
patient succumbed due to progressive disease 110 months after the
initial surgery for gastric ES/PNET.
Discussion
The term ‘Ewing sarcoma’ has been used for tumors
that lack evidence of neuroectodermal differentiation as assessed
by light microscopy, immunohistochemistry and electron microscopy,
whereas the term ‘PNET’ has been employed for tumors that exhibit
neuroectodermal features as evaluated by one or more of these
modalities (8). Originally thought
to be a separate entity, PNET is now known to commonly share
similar immunohistochemical characteristics and an identical
chromosomal translocation t(11;22)(q24;q12) with ES. The two
diseases are now treated as a single entity (8,9). In
the present case, the EWS-FLI1 fusion gene translocation
t(11;22)(q24;q12) indicated that the tumor arising from the
muscularis externa layer of the stomach was a gastric ES/PNET.
The most useful immunohistologic reagent for the
diagnosis of ES/PNETs is a monoclonal antibody to CD99 (HBA/71,
12E7 and 013) that is able to identifty a cell surface protein.
This protein is the product of the MIC2 gene, which is
located on the pseudoautosomal region of the X and Y chromosomes
(1). Immunohistochemical staining
using an antibody to CD99 has been strongly positive in 90 to
nearly 100% of cases reported to be ES/PNETs (10,11).
An evaluation of the five reported cases of gastric ES/PNETs
revealed that tumors from all cases showed positive
immunoreactivity for CD99. Immunohistochemical staining for the
intermediate filament vimentin is usually positive in ES/PNET cells
(1). Similarly, the five reported
cases of gastric ES/PNETs showed positive vimentin expression in
tumor cells. Neural markers, such as S100, chromogranin A,
synaptophysin and neuron-specific enolase, demonstrate variable
immunohistochemical staining in ES/PNET cells, which may be due to
the differing degrees of neuroectodermal differentiation in the
individual tumors (1).
In the five reported cases of gastric ES/PNETs
(Table I), surgical resection was
possible in all cases. Although three of the reported cases,
including our case, had distant metastases, only our patient
underwent multiple repeat metastasectomies for recurrent disease.
Since our patient survived more than 9 years following the initial
surgery for gastric ES/PNETs, repeat metastasectomies for isolated
recurrence, where feasible, may improve survival in patients with
gastric ES/PNETs.
 | Table IClinical data in reported cases of
gastric Ewing sarcoma/primitive neuroectodermal tumors. |
Table I
Clinical data in reported cases of
gastric Ewing sarcoma/primitive neuroectodermal tumors.
| Author | Gender/age
(years) | Tumor size (cm) | Treatment for primary
tumor | Sites of
metastases | Treatment for
metastases | Outcome after
resection of primary tumor |
|---|
| Czekalla et al
(2) | M/14 | 7.0×3.5 | Gastrectomy plus
neoadjuvant and adjuvant chemotherapy | HEP | Systemic
chemotherapy | 24 months; AWD |
| Soulard et al
(3) | F/66 | 8.0×5.0 | Gastrectomy plus
adjuvant chemotherapy | ND | ND | 10 months; DOD |
| Rafailidis et
al (4) | M/68 | 12.0 | Gastrectomy plus
adjuvant chemotherapy | HEP | ND | 13 months; DOD |
| Colovic et al
(5) | F/44 | 6.6×4.6 | Excision | Absent | Absent | 20 months; NED |
| Present case | F/41 | 9.0×5.5 | Gastrectomy
chemotherapy and irradiation | PER, LYM | Metastasectomy plus
systemic | 110 months; DOD |
The current standard chemotherapeutical treatment
for ES/PNETs is a regimen comprising the drugs: vincristine,
doxorubicin, cyclophosphamide, ifosfamide and etoposide.
Chemotherapy often enables the cure of ES/PNET patients with
localized disease. However, in patients with metastatic spread its
benefit is usually limited to extending the length of time of
progression-free survival (12,13).
The findings of Haeusler et al demonstrated that combined
modality treatment (surgery, multi-agent chemotherapy and
radiotherapy) improved survival in patients with metastatic spread
of ES/PNETs (14). The results of
our case demonstrated that combined modality treatment was
effective in achieving long-term survival. These results therefore
indicate the importance of multimodal treatment approaches for
patients with ES/PNETs.
In conclusion, gastric ES/PNETs are rare tumors with
high metastatic potential. Multimodal treatment approaches
including surgery, multi-agent chemotherapy and radiotherapy may
provide a survival benefit for patients with gastric ES/PNETs.
Abbreviations:
|
ES
|
Ewing sarcoma
|
|
PNET
|
primitive neuro-ectodermal tumor
|
|
RECIST
|
Response Evaluation Criteria in Solid
Tumors
|
References
|
1
|
Kempson RL, Fletcher CDM, Evans HL, et al:
Tumors of the Soft Tissues. 3rd edition. The Armed Forces Institute
of Pathology; Washington, DC: pp. 444–452. 2001
|
|
2
|
Czekalla R, Fuchs M, Stölzle A, et al:
Peripheral primitive neuroectodermal tumor of the stomach in a
14-year-old boy: a case report. Eur J Gastroenterol Hepatol.
16:1391–1400. 2004.PubMed/NCBI
|
|
3
|
Soulard R, Claude V, Camparo P, Dufau JP,
Saint-Blancard P and Gros P: Primitive neuroectodermal tumor of the
stomach. Arch Pathol Lab Med. 129:107–110. 2005.PubMed/NCBI
|
|
4
|
Rafailidis S, Ballas K, Psarras K,
Pavlidis T, Symeonidis N, Marakis G and Sakadamis A: Primary Ewing
sarcoma of the stomach – a newly described entity. Eur Surg Res.
42:17–20. 2009.
|
|
5
|
Colovic RB, Grubor NM, Micev MT, Matic SV,
Atkinson HD and Latincic SM: Perigastric extraskeletal Ewing’s
sarcoma: a case report. World J Gastroenterol. 15:245–247.
2009.PubMed/NCBI
|
|
6
|
Obata H, Ueda T, Kawai A, et al: Clinical
outcome of patients with Ewing sarcoma family of tumors of bone in
Japan: the Japanese Musculoskeletal Oncology Group cooperative
study. Cancer. 109:767–775. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Therasse P, Arbuck SG, Eisenhauer EA, et
al: New guidelines to evaluate the response to treatment in solid
tumors. European Organization for Research and Treatment of Cancer,
National Cancer Institute of the United States, National Cancer
Institute of Canada. J Natl Cancer Inst. 92:205–216. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ushigome S, Machinami R and Sorensen PH:
Ewing sarcoma/ Primitive neuroectodermal tumour (PNET). World
Health Organization Classification of Tumours. Pathology and
Genetics of Tumours of Soft Tissue and Bone. Fletcher CDM, Unni KK
and Mertens F: IARC Press; Lyon: pp. 298–300. 2002
|
|
9
|
Skubitz KM and D’Adamo DR: Sarcoma. Mayo
Clin Proc. 82:1409–1432. 2007. View Article : Google Scholar
|
|
10
|
Ambros IM, Ambros PF, Strehl S, Kovar H,
Gadner H and Salzer-Kuntschik M: MIC2 is a specific marker for
Ewing’s sarcoma and peripheral primitive neuroectodermal tumors.
Evidence for a common histogenesis of Ewing’s sarcoma and
peripheral primitive neuroectodermal tumors from MIC2 expression
and specific chromosome aberration. Cancer. 67:1886–1893. 1991.
|
|
11
|
Ramani P, Rampling D and Link M:
Immunocytochemical study of 12E7 in small round-cell tumours of
childhood: an assessment of its sensitivity and specificity.
Histopathology. 23:557–561. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leavey PJ and Collier AB: Ewing sarcoma:
prognostic criteria, outcomes and future treatment. Expert Rev
Anticancer Ther. 8:617–624. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Esiashvili N, Goodman M and Marcus RB Jr:
Changes in incidence and survival of Ewing sarcoma patients over
the past 3 decades: Surveillance Epidemiology and End Results data.
J Pediatr Hematol Oncol. 30:425–430. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haeusler J, Ranft A, Boelling T, et al:
The value of local treatment in patients with primary,
disseminated, multifocal Ewing sarcoma (PDMES). Cancer.
116:443–450. 2010. View Article : Google Scholar : PubMed/NCBI
|