Introduction
Glioblastoma (GBM) is the most frequent type of
glioma, comprising almost 50% of all diagnosed gliomas (1–4). The
median survival of GBM patients remained between 9 and 12 months
until 2004 when patients started chemotherapy with temozolamide
(TMZ). In patients treated with TMZ, the median survival increased
to 14.6 months and the percentage of patients that had a 2-year
survival rate increased to 17%, as compared to that in patients not
treated with TMZ (5). This
improvement in median survival is extremely significant, but
remains modest. The limited success of TMZ in GBM tumors appears to
be related to the occurrence of chemoresistance and to the
inability of TMZ to induce tumor cell death. TMZ is an alkylating
agent that induces the formation of O6-methylguanine in
DNA, which mispairs with thymine during the following cycle of DNA
replication, leading to activation of the apoptotic pathways. In
spite of this mechanism of action, Hirose et al reported
that malignant glioma cells respond to TMZ by undergoing G2/M
arrest and that few glioma cells treated with TMZ underwent
apoptosis (6). In addition, Kanzawa
et al found that TMZ induced autophagy, but not apoptosis in
malignant glioma cells (7).
The mechanisms of TMZ action and the pathways by
which glioma cells escape from death have yet to be adequately
elucidated, and the reduced efficacy of TMZ in GBM treatment has
yet to be determined. The reduced efficacy of TMZ in gliomas was
initially attributed to the activity of MGMT which removes the DNA
adducts. However, Hegi et al showed that even when the MGMT
promoter was methylated, the median survival was 21.7 months
(8). These results indicate that
the mechanism of TMZ action may be overlapped by the survival
signaling pathways. Previous studies reported that in patient tumor
tissue samples the ERK1/2 and Pi3K/Akt were phosphorylated,
indicating that these survival pathways were active in glioma cells
(7,9–12).
Since these signaling pathways sustain key features that
characterize gliomas, e.g., enhance proliferation and invasion,
protect from proapoptotic stimuli and activate autophagy, it is
likely that they may contribute to chemoresistance.
The activation status of cell survival pathways
Pi3K/Akt, ERK1/2 and of autophagy in GBM cells treated with TMZ is
poorly understood. Since TMZ is the first-line treatment in
patients with GBM and 45% of GBM patients are resistant to
TMZ-treatment, the purpose of this study was to evaluate whether
the activation status of Pi3K/Akt, ERK1/2 and autophagy interferes
with the mechanism of action of TMZ.
Materials and methods
Reagents
DMEM, fetal bovine serum (FBS), propidium iodide
(PI) and Hoechst were supplied by Invitrogen (Paisley, UK). The
protease and phosphatase inhibitors were supplied by Roche
(Indianapolis, IN, USA). Antibodies for Phospho-Akt, Phospho-ERK1/2
and total ERK1/2 were purchased from Cell Signalling Technology
(MA, USA), the LC3 antibody was purchased from Affinity Bioreagents
(Rockford, IL, USA) and mouse anti-actin antibody was purchased
from Boehringer Mannheim (Germany). The phosphatase linked
anti-mouse and anti-rabbit antibodies, and the substrate for the
phosphatase were obtained from GE Healthcare (UK). PVDF membranes
were purchased to Millipore (MA, USA). TMZ and the other chemicals
were purchased from Sigma Chemicals (St. Louis, MO, USA). TMZ was
dissolved in dimethyl sulfoxide (DMSO) at a concentration stock of
0.133 M. This stock was aliquoted and diluted with culture medium
according to the used concentration. The bromodeoxyuridine (BrdUrd)
kit to detect cell proliferation was purchased from Roche.
Cell line and cell culture
conditions
The U-118 GBM cell line was purchased from the
American Tissue Culture Collection, and maintained in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 3.5 mg/ml glucose,
0.1 mg/ml penicillin, 0.14 mg/ml streptomycin and 10% inactivated
FBS. The cultured cells were maintained at 37°C, in an atmosphere
containing 95% air and 5% CO2. Cells were subcultured
every 48 h by lifting them up with a cell scrapper. The cells were
then centrifuged and resuspended in fresh DMEM. For the
experiments, unsynchronized cells were treated with different
concentrations of TMZ (0, 10, 20, 100, 250 and 500 μM) for 24 and
48 h. Assays were previously performed in the presence of DMSO,
which corresponded to each TMZ concentration.
Cell proliferation assay
Cells were plated in 96 multi-well plates at
different TMZ concentrations, for 24 and 48 h. The effect of TMZ on
the proliferation rate was assayed by a BrdUrd kit. Briefly, at the
end of the incubation period 10 μl of a BrdUrd solution in culture
medium (1:100) was added to each well, and the cells were incubated
for 6 h. The culture medium was then removed by rapidly flicking it
off the multi-well plate. The cells were fixed, incubated with an
anti-BrdU antibody linked to a peroxidase and then incubated with a
peroxidase substrate. The absorbance of the final solution was read
in an ELISA plate reader at a 450-nm wavelength.
Cell cycle analysis by flow
cytometry
Cells were plated in six multi-well plates and
incubated with TMZ, wortmannin, U-0126 and rapamycin for 24 and 48
h. At the end of the incubation period, cells were centrifuged at
1,500 rpm for 10 min, the culture medium was discarded and the
pellet was resuspended in a solution of 70% ethanol and maintained
overnight. The cells were then centrifuged at 1,500 rpm for 10 min,
the pellet was resuspended and incubated for 1 h in the dark, at
room temperature, in a solution of PBS containing 10 μl/ml PI and
10 μl/ml RNAse (13). The PI
fluorescence was measured on a FACScan flow cytometer (BD
FACSCalibur™) and the data were gated to exclude cell debris and
aggregates. Data were analyzed on WinMDI for Windows.
Staining of U-118 TMZ-treated cells with
Hoescht 33258
Cells were plated in six multi-well plates and
incubated with TMZ for 24 and 48 h. After that, cells were washed
in a PBS solution, centrifuged at 1,500 rpm for 10 min and
incubated for 15 min with a solution of methanol and acetone (1:1).
Cells were then centrifuged at 1,500 rpm for 10 min, washed with
PBS and incubated with 5 μg/ml Hoescht 33258 solution for 5 min at
room temperature. After the incubation time, cells were washed and
resuspended in PBS and mounted with Vectashield on glass slides.
The images were captured under a Zeiss LSM 510 Meta confocal
microscope at a magnification of ×63, and viewed on a Zeiss LSM
image browser (Version 4.2.0.121; Carl Zeiss Inc., Germany).
Study of protein expression
Cells incubated with TMZ for 24 and 48 h were
centrifuged at 1,500 rpm for 10 min at 4°C. The supernatant was
discarded, cells were treated with RIPA buffer (50 mM Tris HCl at
pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS
and 2 mM EDTA) supplemented with protease, phosphatase inhibitors
and DTT, and cell suspensions were sonicated. The protein content
of each sample was assessed and then proteins were denatured.
Following that, the buffer (Tris 0.5 mM, pH 6.8; 50% glycerol, 10%
SDS, 10% 2β-mercaptoethanol and blue bromophenol) was added to the
samples at a 1:1 ratio (14).
Protein extracts were then boiled at 95°C for 5 min and used.
For Western blotting, 70 μg of protein was separated
on a 12% SDS-PAGE and then transferred to a PVDF membrane. The PVDF
membrane was blocked with a solution of 5% milk in TBST for 1 h at
room temperature and incubated overnight at 4°C with the primary
antibody against LC3, p-Pi3K/Akt or p-ERK1/2 diluted in TBST with
1% milk supplemented with azide. Bound antibody was detected with
an alkaline phosphatase conjugated anti-rabbit antibody, using
enhanced chemifluorescence detection reagents. The protein
expression was quantitated using the ImageQuant TL for Windows
(version 2005; Amersham Biosciences, Piscataway, NJ, USA) with the
expression of β-actin as a loading control for LC3, as well as
total Pi3K/Akt and total ERK1/2 as a loading control for p-Pi3K/Akt
and p-ERK1/2, respectively.
Statistical analysis
Statistical analysis was performed on GraphPad Prism
5 for Windows (version 5.00; GraphPad Software, Inc., San Diego,
CA, USA). Statistical significance within groups was assessed by a
t-test and between groups by a two-way ANOVA, with a significance
threshold of p≤0.05.
Results
TMZ reduced U-118 proliferation
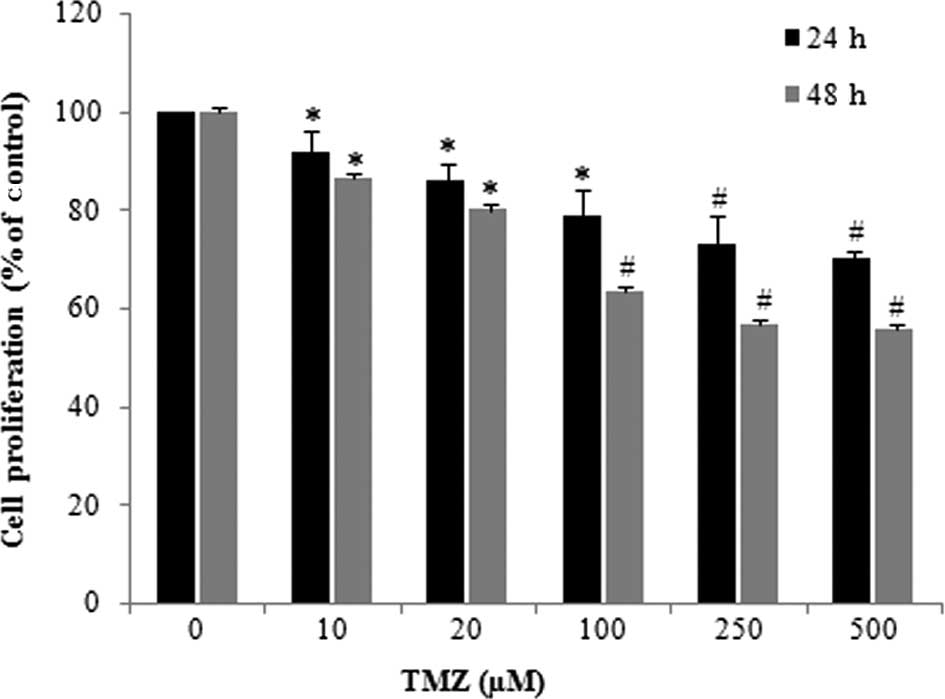
The effect of TMZ in U-118 cell proliferation was
evaluated by quantification of the incorporated BrdUrd. TMZ
treatment induced a significant inhibition of the BrdUrd
incorporation in glioma cells, which occurred in a dose- and
time-dependent manner (Fig. 1). The
effect of TMZ was particularly evident when U-118 cells were
incubated with TMZ concentrations of >100 μM. When U-118 cells
were incubated with TMZ (250 μM) the proliferation rate was
inhibited by 43.2%, as compared to that observed in the control
cells.
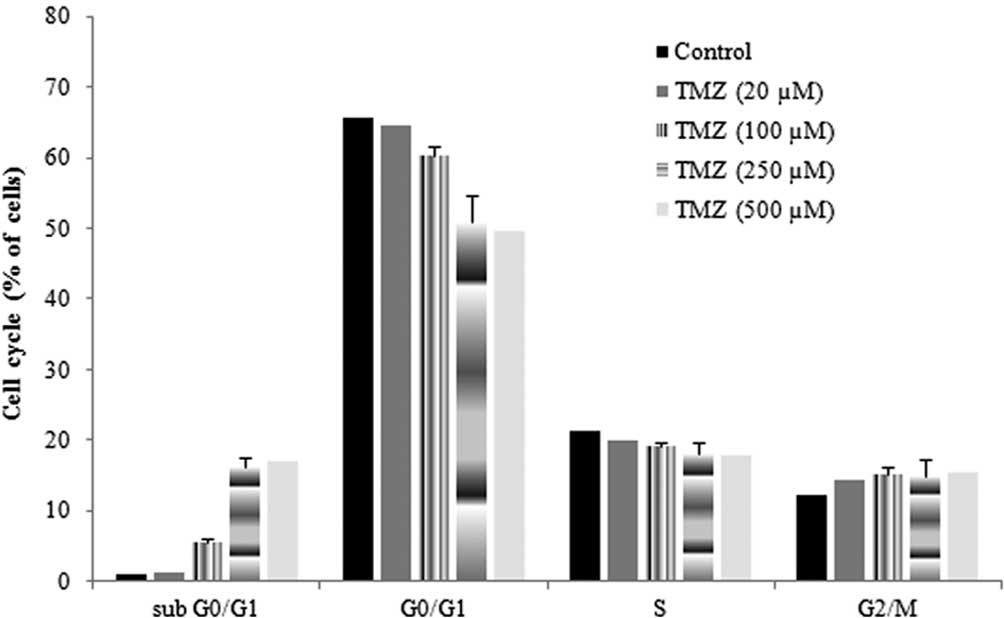
Effect of TMZ on the cell cycle
To determine whether the reduced proliferation was
related to an increase in apoptosis, the effect of TMZ in the cell
cycle was also evaluated. The cell cycle of untreated cells was
characterized by a long and well-defined G0/G1 peak, with a
slightly prominent G2/M peak and a relatively low apoptotic
fraction. When cells were incubated with TMZ, the percentage of
cells in G0/G1 was reduced, the percentage of cells in sub G0/G1,
which were considered as apoptotic cells, significantly increased
and the percentage of cells in S and G2/M was not significantly
altered (Fig. 2). These alterations
were more evident when cells were incubated for 48 h with TMZ. In
the presence of TMZ (250 μM), the percentage of cells in the sub
G0/G1 phase increased 15.4% and the percentage of cells in G0/G1
decreased 14.6%, as compared to the control. These results
indicated that TMZ had a reduced ability to induce apoptosis and
also did not induce cell-cycle arrest in G1/S or in G2/M.
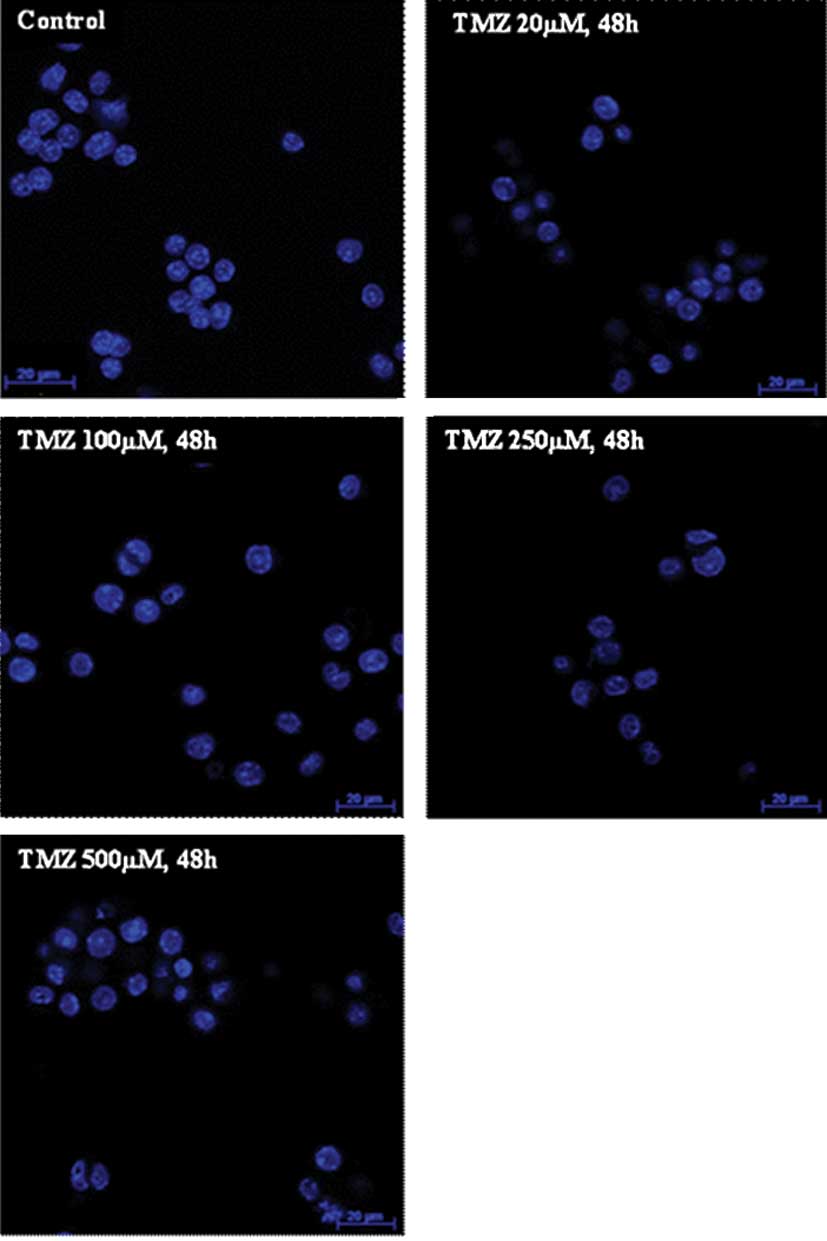
TMZ induced chromatin condensation
To evaluate whether TMZ induced morphological
alterations compatible with apoptosis, U-118 cells were stained
with Hoechst 33258 dye. When cells were incubated with TMZ, it was
possible to detect chromatin condensation, irregular nuclei contour
and pycnotic nuclei in certain cells confirming the results
obtained from cell cycle analysis (Fig.
3). These observations were more evident when the TMZ
concentration was >100 μM, and also when cells were incubated
with TMZ for 48 h.
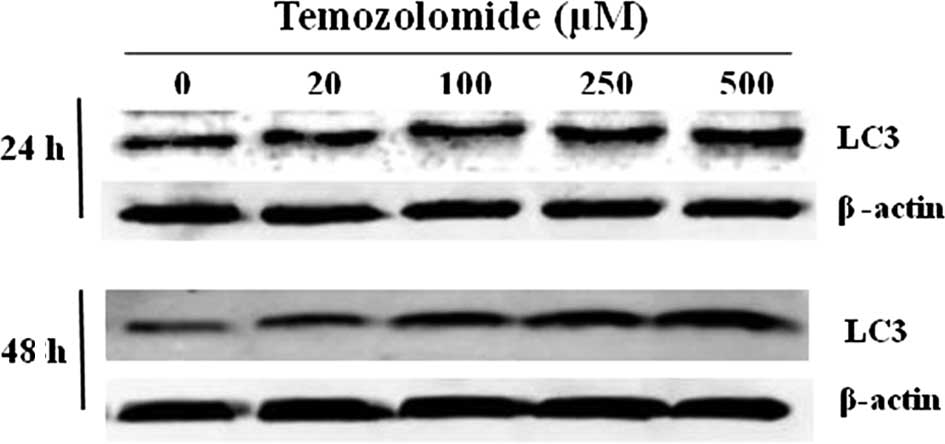
Expression of LC3 in the presence of
TMZ
Since there was a significant decrease in U-118 cell
proliferation and the percentage of apoptotic cells was reduced,
the expression of LC3, one of the autophagosome-membrane proteins,
was analyzed. The results showed that U-118 cells constitutively
expressed the LC3 protein. However, in U-118 cells incubated with
TMZ there was an increase in the LC3 expression, which reached a
maximum in the presence of 500 μM of TMZ. This increase in LC3
expression was statistically significant when cells were incubated
with TMZ for 48 h in the presence of 250 and 500 μM (Fig. 4). These results may justify the
reduction of proliferation in U-118 glioma cells incubated with
TMZ, as well as the development of chemoresistance.
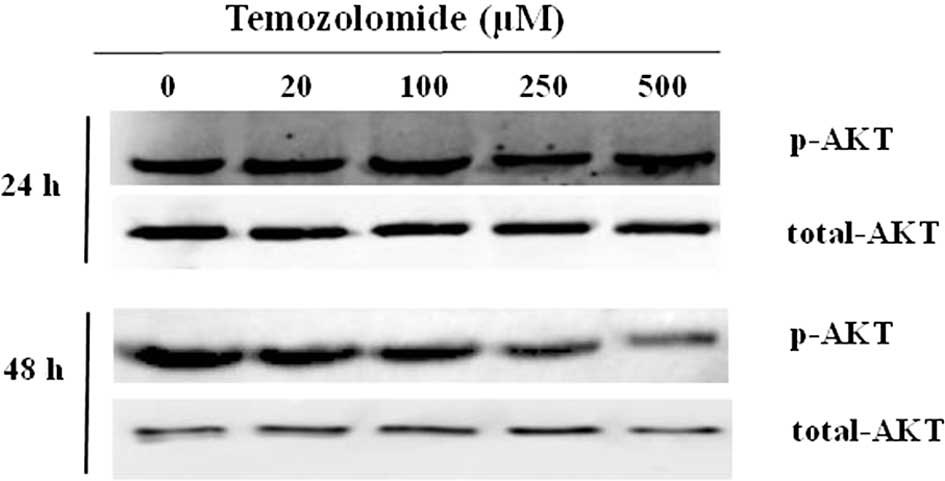
Role of Pi3K/Akt and ERK1/2 MAP kinase on
U-118 cell survival
Since the induction of apoptosis is dependent on the
DNA repair systems, as well as on the activity of various signaling
pathways, the phosphorylation status of Pi3K/Akt and ERK1/2 MAP
kinase was further evaluated by Western blotting. The results
showed that the endogenous Akt was characterized by a basal
phosphorylation of the Ser473 (Fig.
5). The basal level of p-Akt was maintained when cells were
incubated for 24 h, and it was slightly reduced when cells were
incubated for 48 h with 250 and 500 μM of TMZ. This reduction was
not statistically significant and was probably associated with the
increased apoptosis rate observed in the presence of TMZ (250 and
500 μM) for 48 h.
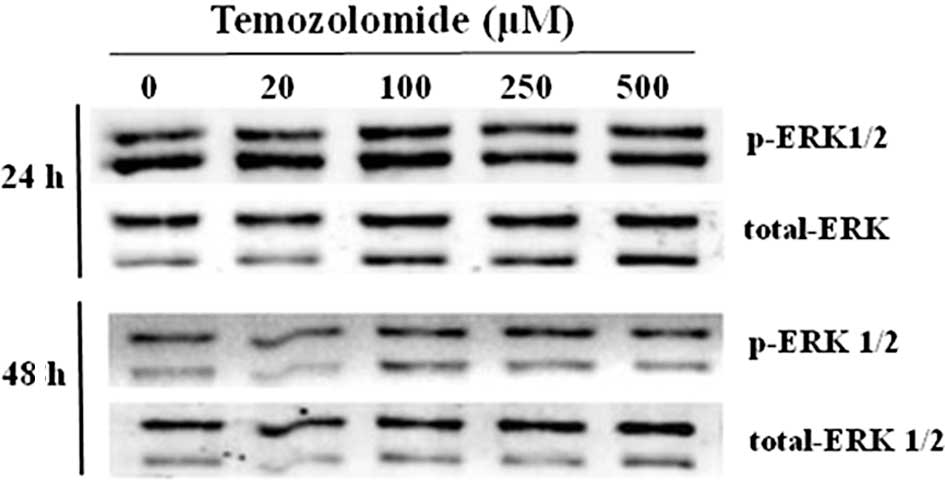
The activation status of ERK1/2 MAP kinase was
evaluated by determining the content of the p-ERK1/2 by Western
blotting. The results showed that there was a basal activation of
ERK1 and 2 in the U-118 glioma cells (Fig. 6). In the presence of TMZ, the amount
of p-ERK1/2 was not significantly altered.
Induction of apoptosis in U-118
cells
Considering that the Pi3K/Akt and ERK1/2 MAP kinase
were maintained in the phosphorylated forms in the presence of TMZ
(250 μM), and that these signaling pathways stimulate cellular
proliferation, U-118 cells were incubated with wortmannin, the
inhibitor of Pi3K/Akt; U-0126, the inhibitor of ERK1/2 MAP kinase
and rapamycin, the inhibitor of mTOR. Cell cycle was then studied
by flow cytometry, 48 h after the addition of the inhibitors, and
the percentage of cells in the sub G0/G1 phase was quantified
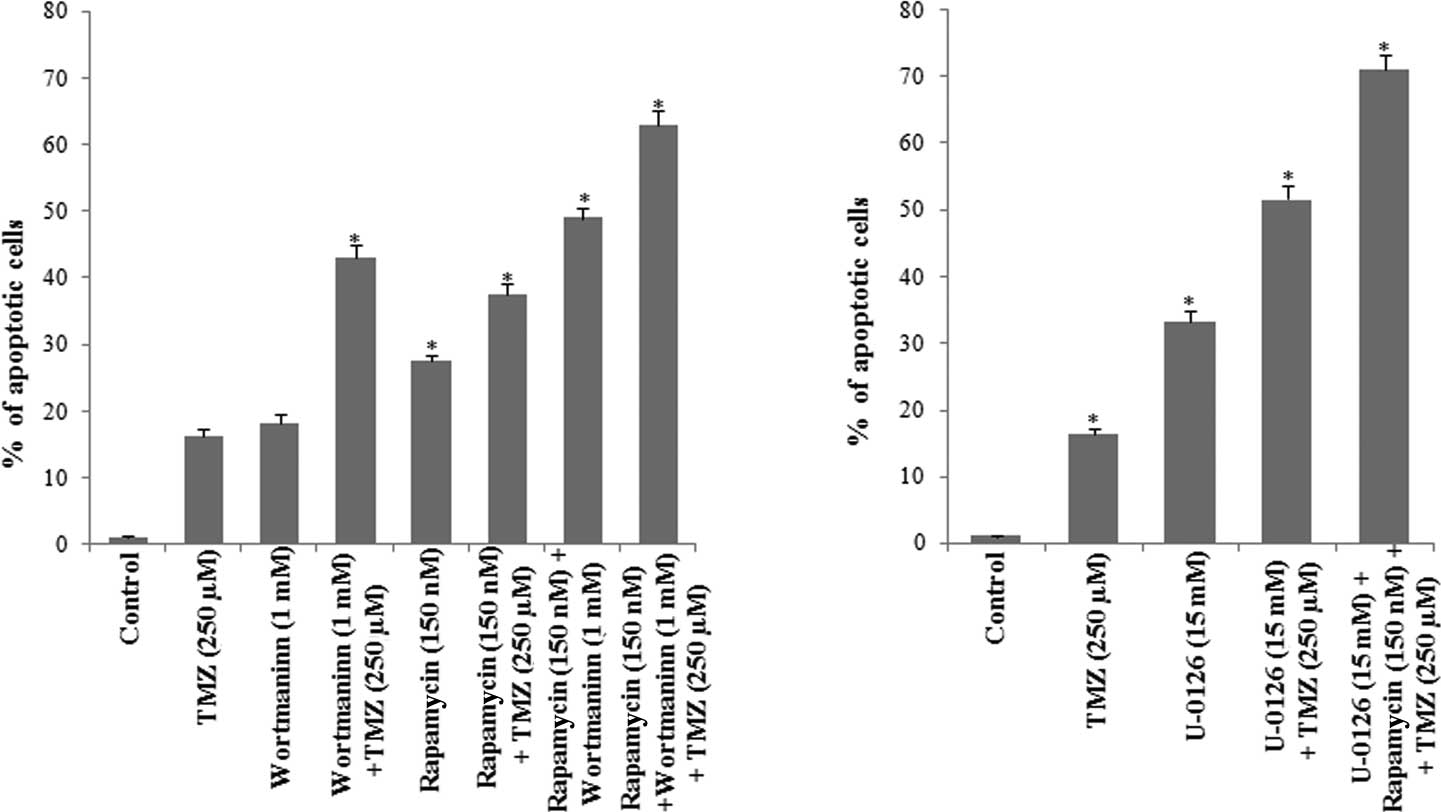
(Fig. 7).
The results showed that the percentage of apoptotic
cells in the presence of wortmannin was 18.24%, which is similar to
that observed in the presence of TMZ. However, when wortmannin was
used in combination with TMZ, the percentage of apoptotic cells was
43.09%, indicating that the inhibition of Pi3K/Akt contributes to
the chemoresistance of glioma cells to TMZ.
When cells were incubated with rapamycin, the
percentage of apoptotic cells increased to 26.5% as compared to
that observed in the control cells, and was 11.13% higher than the
percentage of apoptosis detected in the presence of TMZ. When cells
were simultaneously incubated with rapamycin and TMZ, the
percentage of apoptotic cells was 37.44%, indicating that mTOR also
contributes to U-118 cell survival.
Since mTOR is a downstream effector of Akt, U-118
cells were further incubated with wortmannin, rapamycin and TMZ. As
shown in Fig. 7A, the percentage of
apoptotic cells was found to be 63.01%, indicating that the
Pi3K/Akt/mTOR signaling pathways contribute to the chemoresistance
of these glioma cells.
The contribution of ERK1/2 MAP kinase to glioma cell
survival was evaluated in the presence of the U-0126. As shown in
Fig. 7B, the percentage of
apoptotic cells was 33.3% in the presence of U-0126. When U-0126
was combined with TMZ the percentage of apoptotic cells was 51.6%,
an increase of 35.25% as compared to the percentage of apoptotic
cells detected in the presence of TMZ (Fig. 7).
U-118 cells were then incubated with wortmannin,
rapamycin, U-0126 and TMZ, and the cell cycle was evaluated. The
percentage of apoptotic cells determined in this condition was
71.0%, indicating that the survival of glioma cells depends on the
activity of Akt/mTOR and ERK1/2 MAP kinase, and also that the
blockage of the two signaling pathways contributes to an increase
in the efficacy of TMZ.
Discussion
The EORTC/NCIC trial demonstrated an improvement in
overall median survival of GBM patients from 12.1 months in
patients submitted to radiotherapy, to 14.6 months in the
TMZ-treated patients (15–18). This survival increment was extended
by 6.4 months when the promoter of the MGMT was methylated
(8,11). As a consequence of these results,
TMZ is considered the gold standard in GBM treatment. However, the
increment in survival remains reduced and therefore the study of
the mechanism of action of TMZ is a fundamental issue.
The ability of alkylating agents to induce apoptosis
is dependent not only on the MGMT activity, but also on the
activity of various survival pathways. Thus, analysis of the TMZ
effect on apoptosis, as well as the study of the interactions
between TMZ and the survival pathways (Pi3K/Akt, ERK1/2 and
autophagy), may contribute to improving the survival of GBM
patients.
Regarding the mechanism of TMZ action, the results
from previous studies were contradictory. Kanzawa et al
showed that TMZ induced autophagy, but not apoptosis in malignant
glioma cells (7). On the other
hand, Hirose et al reported that in glioma cells TMZ induced
a low level of apoptosis as compared to that observed in lymphoid
cells, and that TMZ induced a cell cycle arrest in G2/M (19). In addition, Roos et al showed
that cell death induced by TMZ in gliomas was due to apoptosis
(20).
In our study, TMZ induced a significant increase in
the expression of LC3, the protein associated with autophagosomes.
The activation of autophagy may explain the reduction in U-118
proliferation observed in the presence of TMZ and may also
constitute a survival mechanism of glioma cells. The signaling
pathway of autophagy remains to be elucidated, since it may allow
for the adaptation of tumor cells to adverse conditions induced by
cancer therapy, contributing to tumor cell survival and
consequently to chemoresistance (21–24).
However, when irreversibly activated, autophagy triggers cell
death, contributing to an increase in therapeutic efficacy
(21–25). Therefore, understanding the
mechanism involved in the activation of irreversibe autophagy may
contribute to reducing the survival of glioma.
Our study also demonstrated that TMZ induced a low
level of apoptosis that was not accompanied by cell cycle arrest.
The reduced level of apoptosis may be associated with the activity
of MGMT, Pi3K/Akt and/or ERK1/2 MAP kinase. Since the U-118 glioma
cells are characterized by hypermethylation of the MGMT promoter
(26), the reduced ability of TMZ
to induce apoptosis does not appear to be associated with the
activity of this DNA repair enzyme. Therefore, we analyzed the
phosphorylation of the signaling pathways Pi3K/Akt and ERK1/2 MAP
kinase.
The results indicated that Pi3K/Akt was
constitutively active in the U-118 cells and that the active state
was maintained in glioma cells treated with TMZ. Since a permanent
phosphorylation of Pi3K/Akt may suppress the G2 checkpoint by
altering activation of the DNA damage signal transducer Chk2 and
the downstream effectors of the G2 checkpoint (12), we suggest that the active state of
Pi3K/Akt contributed to the ability of U-118 cells to overcome the
G2 checkpoint, thereby avoiding the effect of TMZ. Confirming the
suppression of G2/M checkpoint is the fact that we did not observe
any accumulation of cells in G2/M when cells were incubated with
TMZ. To confirm the contribution of Pi3K/Akt to chemoresistance, we
incubated U-118 cells with wortmannin, a specific inhibitor of
Pi3K/Akt, and measured apoptosis. In the presence of wortmannin the
percentage of apoptotic cells increased 17.24% as compared to the
control cells, indicating that wortmanin induced a similar
percentage of apoptosis as TMZ. However, in the presence of
wortmannin and TMZ the percentage of apoptotic cells was 43.01%, an
increase of 26.7% as compared to cells treated with TMZ, confirming
that Pi3K/Akt contributed to the survival and chemoresistance of
U-118 cells to TMZ. Since wortmannin inhibits the activity of mTOR
and previous results showed that proliferation associated with the
activity of Pi3K/Akt signaling pathway is mediated by mTOR
(27,28), we evaluated the effect of rapamycin,
the inhibitor of mTOR, on U-118 cell survival.
The results show that mTOR plays a crucial role in
glioma cell survival, since the percentage of apoptosis was 26.5%
in the presence of rapamycin. We also showed that the percentage of
apoptotic cells increased to 37.44% when rapamycin was combined
with TMZ, indicating the existence of a synergistic effect. In
addition, we reported that the dual inhibition of Pi3K/mTOR in the
presence of TMZ renders cells more susceptible to apoptosis and
therefore may constitute a therapeutic target.
Regarding the study of ERK1/2 MAP kinase activation,
our results indicated that U-118 cells were characterized by a
constitutive activation of this signaling pathway which was
maintained in the presence of TMZ. To evaluate the contribution of
ERK1/2 MAP kinase to U-118 survival, cells were incubated with the
inhibitor U-0126 in the absence and presence of TMZ. The results
showed that in the presence of the ERK1/2 MAP kinase inhibitor, the
percentage of apoptotic cells was 33.3%. When cells were treated
with U-0126 combined with TMZ, the percentage of apoptotic cells
was 51.6%, indicating that this signaling pathway also contributes
to the survival of U-118 cells.
Previous studies reported that the sustained
activation of ERK1/ERK2 contributed to G1- to S-phase progression
and protected cells from apoptotic signaling (29,30).
Taking into consideration that the activation of Pi3K/Akt inhibits
the G2-M arrest (6,12) and that ERK1/2 MAP kinase inhibits
the arrest in G0/G1 (18,19), we hypothesized that in order to
induce GBM cell death the two signaling pathways should be blocked.
In the presence of wortmannin, rapamycin, U-0126 and TMZ the
percentage of apoptosis increased to 71.01%, which evidences the
existence of a synergistic effect between the survival pathways and
TMZ. Our results emphasize the plasticity of glioma cells and their
ability to override difficulties associated with the blocking of
signaling pathways. This plasticity may justify the resistance of
glioma cells to TMZ as well as the differences in GBM cell
sensitivity to this alkylating agent. These results also suggest
the need to combine traditional chemotherapy with molecular-based
therapy, in particular therapy associated with signaling
pathways.
In conclusion, our results indicate that TMZ by
itself is not effective in inducing cell death, even in the U-118
cell line, which is hypermethylated. To overcome the resistance of
U-118 cells to TMZ it appears essential to evaluate i) the
activation status of the Pi3K/Akt, mTOR, ERK1/2 MAP kinase and
autophagy, and ii) the synergistic effect between TMZ and the
inhibitors of these signaling pathways. Therefore, we hypothesize
that before glioma patients initiate TMZ therapy, it is necessary
to evaluate the phosphorylation of the Pi3K/Akt and ERK1/2 MAP
kinase and prevent the activation of autophagy by TMZ.
References
|
1
|
Schwartzbaum JA, Fisher JL, Aldape KD and
Wrensch M: Epidemiology and molecular pathology of glioma. Nat Clin
Pract Neurol. 2:494–503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohgaki H and Kleihues P: Epidemiology and
etiology of gliomas. Acta Neuropathol. 109:93–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
CBTRUS. Statistical Report: Primary Brain
Tumors in the United States, 2004—2006. Central Brain Tumor
Registry of the United States; pp. 1–65. 2010
|
|
4
|
Hadjipanayis CG and van Meir EG: Brain
cancer propagating cells: biology, genetics and targeted therapies.
Trends Mol Med. 15:519–530. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirose Y, Berger MS and Pieper RO:
Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates
temozolomide-induced toxicity in a p53-independent manner in human
glioblastoma cells. Cancer Res. 61:5843–5849. 2001.
|
|
7
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hegi M, Diserens A, Gorlia T, et al: MGMT
gene silencing and benefit from temozolomide in glioblastoma. N
Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Keating AK, Kim GK, Jones AE, Donson AM,
Ware K, Mulcahy JM, Salzberg DB, Foreman NK, Liang X, Thorburn A
and Graham DK: Inhibition of Mer and Axl receptor tyrosine kinases
in astrocytoma cells leads to increased apoptosis and improved
chemosensitivity. Mol Cancer Ther. 9:1298–1307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akhavan D, Cloughesy TF and Mischel PS:
mTOR signaling in glioblastoma: lessons learned from bench to
bedside. Neuro Oncol. 12:882–889. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mrugala MM and Chamberlain MC: Mechanisms
of disease: temozolomide and glioblastoma – look to the future. Nat
Clin Pract Oncol. 5:476–486. 2008.
|
|
12
|
Hirose Y, Katayama M, Mirzoeva OK, Berger
MS and Pieper RO: Akt activation suppresses Chk2-mediated,
methylating agent-induced G2 arrest and protects from
temozolomide-induced mitotic catastrophe and cellular senescence.
Cancer Res. 65:4861–4869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carmo A, Patricio I, Cruz MT, Carvalheiro
H and Lopes MC: CXCL12/CXCR4 promotes motility and proliferation of
glioma cells. Cancer Biol Ther. 9:56–65. 2010. View Article : Google Scholar
|
|
14
|
Neves BM, Cruz MT, Francisco V, Goncalo M,
Figueiredo A, Duarte CB and Lopes MC: Differential modulation of
CXCR4 and CD40 protein levels by skin sensitizers and irritants in
the FSDC cell line. Toxicol Lett. 177:74–82. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Van Meir EG, Hadjipanayis CG, Norden AD,
Shu H, Wen PY and Olson JJ: Exciting new advances in
neuro-oncology: the avenue to a cure for malignant glioma. CA
Cancer J Clin. 60:166–193. 2010.PubMed/NCBI
|
|
16
|
Sathornsumetee S, Reardon DA, Desjardins
A, Quinn JA, Vredenburgh JJ and Rich JN: Molecularly targeted
therapy for malignant glioma. Cancer. 110:13–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Furnari F, Fenton T, Bachoo R, et al:
Malignant astrocytic glioma:genetics, biology, and paths to
treatment. Genes and Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gladson CL, Prayson RA and Liu WM: The
pathobiology of glioma tumors. Annu Rev Pathol Mech Dis. 5:33–50.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hirose Y, Berger MS and Pieper RO: p53
effects both the duration of G2/M arrest and the fate of
temozolomide-treated human glioblastoma cells. Cancer Res.
61:1957–1963. 2001.
|
|
20
|
Roos WP, Batista LF, Naumann SC, Wick W,
Weller M, Menck CF and Kaina B: Apoptosis in malignant glioma cells
triggered by the temozolomide-induced DNA lesion O6-methylguanine.
Oncogene. 26:186–197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ogier-Denisy E and Codogno P: Autophagy: a
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
22
|
Mariño G and López-Otín C: Autophagy:
molecular mechanisms, physiological functions and relevance in
human pathology. Cell Mol Life Sci. 61:1439–1454. 2004.PubMed/NCBI
|
|
23
|
Fu J, Shao C, Chen F, Ng H and Chen Z:
Autophagy induced by valproic acid is associated with oxidative
stress in glioma cell lines. Neuro Oncol. 12:328–340. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thorburn A: Apoptosis and autophagy:
regulatory connections between two supposedly different processes.
Apoptosis. 13:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Katayama M, Kawaguchi T, Berger MS and
Pieper RO: DNA damaging agent-induced autophagy produces a
cytoprotective adenosine triphosphate surge in malignant glioma
cells. Cell Death Differ. 14:548–558. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lorente A, Mueller W, Urdangarín E, Lázcoz
P, Deimling A and Castresana JS: Detection of methylation in
promoter sequences by melting curve analysis-based semiquantitative
real time PCR. BMC Cancer. 8:61–75. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ronellenfitsch MW, Brucker DP, Burger MC,
Wolking S, Tritschler F, Rieger J, Wick W, Weller M and Steinbach
JP: Antagonism of the mammalian target of rapamycin selectively
mediates metabolic effects of epidermal growth factor receptor
inhibition and protects human malignant glioma cells
fromhypoxia-induced cell death. Brain. 13:1509–1522. 2009.
View Article : Google Scholar
|
|
28
|
Law BK: Rapamycin: an anti-cancer
immunosuppressant? Crit Rev Oncol Hematol. 56:47–60. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meloche S and Pouyssegur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tran SE, Holmstrom TH, Ahonen M, Kahari VM
and Eriksson JE: MAPK/ERK overrides the apoptotic signaling from
Fas, TNF, and TRAIL receptors. J Biol Chem. 276:16484–16490. 2001.
View Article : Google Scholar : PubMed/NCBI
|