Introduction
Polycyclic aromatic hydrocarbons (PAHs) are widely
distributed in nature and are considered to be significant
environmental carcinogens. Although they are generally formed at
high temperatures, PAHs should be considered as biologically
inactive. The major sources of these hydrocarbons include, but are
not limited to, power plants, gasoline, coal tar and diesel
engines. PAHs are soluble in lipid, can be metabolized and are
capable of interacting with cellular components, such as protein
and nucleic acid. The metabolic activation of PAH is responsible
for their carcinogenic properties. Since PAHs require metabolic
activation, they are considered to be indirect acting carcinogens
(1). On the other hand, DNA
intercalators, an important class of antitumoral DNA binders, are
characterized by the insertion of planar aromatic or heteroaromatic
rings between DNA base pairs (2,3), as in
the case of anthracyclines, acridines and ellipticines (4,5), which
are thought to poison topoisomerases I and II (6). Various factors are involved in the
stabilization of the drug-DNA complex, of which hydrogen bonding
and π-stacking are the most important. Moreover, PAHs constitute an
important class for the design of new chemotherapeutic DNA
intercalators (7,8).
PAH-containing anticancer compounds were first
reported to be present in either the anthracene (8–11) or
the pyrene ring systems (1,12–14).
DNA-binding molecules are considered to be an important class of
drugs in anticancer therapy (15).
Although it is well-established that DNA binding is not sufficient
to confer cytotoxic activities, interaction with DNA is often
considered a necessary criterion for maintaining a cytotoxic
effect. The antitumoral anthracyclines daunorubicin and doxorubicin
and the synthetic anthracene-9,10-dione mitoxantrone are potent
agents in clinical use at present, with broad application in the
treatment of several leukemias and lymphomas as well as in
combination chemotherapy of solid tumors (16,17).
It is well-known that cancer is the leading cause of
death in people under the age of 85 in the United States, and
mortality from this disease appears to be on the increase (18). Therefore, it is imperative to
develop new compounds and strategies to decrease the incidence and
mortality of cancer. As a part of our ongoing research (7,19–29) to
synthesize new PAH-bearing anticancer agents, we report the
synthesis and in vitro cytotoxic evaluation of certain new
PAH compounds (Fig. 1).
Materials and methods
Materials
The following materials were obtained for the study:
PAH (chrysene and pyrene), bismuth nitrate pentahydrate,
montmorillonite KSF clay, indium, ammonium chloride, isobutyl
chloroformate, 1.0 M borane in tetrahydrofuran, reagent grade
solvents (Aldrich Chemical Co., St. Louis, MO, USA),
dimethylsulfoxide (DMSO; Sigma-Aldrich Corp., St. Louis, MO, USA),
phosphate-buffered saline (PBS), Dulbecco's modified Eagle's medium
(DMEM) (Invitrogen, Carlsbad, CA, USA), fetal bovine serum (FBS;
Invitrogen), McCoy's media (Invitrogen) and doxorubicin (Fisher
Scientific, Pittsburgh, PA, USA).
Synthesis of the compounds 1, 2 and
3
Melting points were determined in a Fisher
Scientific electrochemical Mel-Temp* manual melting point apparatus
(Model 1001) equipped with a 300°C thermometer. Elemental analyses
(C, H, N) were conducted using the Perkin-Elmer 2400 series II
elemental analyzer. Their results were found to be in agreement
(±0.2%) with the calculated values for C, H, N. FT-IR spectra were
registered on a Bruker IFS 55 Equinox FT-IR spectrophotometer as
KBr discs. 1H-NMR (300 MHz) and 13C-NMR (75.4
MHz) spectra were obtained at room temperature with JEOL
Eclipse-300 equipment using TMS as an internal standard and
CDCl3 as a solvent. Analytical grade chemicals
(Sigma-Aldrich Corp.) were used throughout the study. Deionized
water was used for the preparation of all aqueous solutions.
PAH nitration
Chrysene or pyrene (1 mmol) and montmorillonite KSF
clay (500 mg; Aldrich Chemical Co.) were added to a suspension of
bismuth nitrate pentahydrate (1 mmol) in anhydrous dichloromethane
(10 ml). The solvent was then evaporated under reduced pressure and
the reaction mixture was irradiated in a kitchen microwave for 6
min (2×3 min). After every 2 min the reaction was monitored by TLC.
After completion of the reaction, the reaction mixture was washed
with dichloromethane (3×5 ml) and the solvent was evaporated by
reduced pressure distillation. The pure product was isolated by
crystallization from a dichloromethane/hexanes mixture in excellent
yield (>90%).
Reduction of the nitrated PAH to the
corresponding amine
The nitrated PAH (1 mmol) and indium (570 mg), 2.5
ml ethanol and 2.5 ml 20% aqueous ammonium chloride solution were
refluxed vigorously for 24 h (monitored by TLC). After completion
of the reaction, the mixture was filtered through a Büchner funnel
and the filtrate was extracted with dichloromethane (2×3 ml). The
dichloromethane layer was washed with brine and water successively
and dried over anhydrous sodium sulfate. The pure amine was
isolated by crystallization from a dichloromethane/hexanes mixture
in excellent yield (>80%).
Coupling of the polyaromatic amines with
isobutyl chloro-formate
The polyaromatic amine (1 mmol) was stirred with
triethylamine (3 mmol) in anhydrous dichloromethane (5 ml) at a
temperature of −5 to 0°C, (1.8 mmol in 2 ml anhydrous
dichloromethane) was then added dropwise. Following the addition of
Isobutyl chloroformate, the mixture was agitated for 24 h
(monitored by TLC). After completion of the reaction, the mixture
was washed with a saturated solution of sodium bicarbonate, brine
and water successively. The pure product was isolated after column
chromatography over silica gel (>70% yield).
Reduction of 2 to 3
Compound 2 (1 mmol) was refluxed with 6 ml of 1.0 M
borane/tetrahydrofuran solution for 36 h. Then, 5 ml of 4%
hydrochloric acid solution was added and the mixture was again
refluxed for another 24 h. After completion of the reaction, the pH
of the solution was maintained at ~7.0 by 10% aqueous sodium
hydroxide solution and the mixture was extracted with ethyl acetate
(3×3 ml). The organic layer was washed with brine and water
successively. The pure product (compound 3) was isolated after
column chromatography over silica gel (78% yield).
Isobutyl chrysen-6-ylcarbamate (compound
1)
mp 181°C; IR (KBr disc, cm−1): 3278,
3269, 1695, 1535, 1239, 1165, 972, 829; 1H-NMR δ
(CDCl3, ppm): 0.99 (m, 6H, methyl), 2.00 (m, 1H,
methine), 3.98 (m, 2H, methylene), 7.19 (broad s, NH), 7.89–8.09
(m, 11H, Ar-H); 13C-NMR (CDCl3, ppm) δ: 19.56
(2C), 28.40, 70.76, 111.98, 119.78, 122.43, 122.47, 126.54, 126.87,
126.89, 126.94, 126.98, 127.07, 127.98, 128.65, 128.69, 129.87,
129.98, 130.08, 131.86, 131.99, 156.51. Anal. Calcd. for
C23H21NO2: C, 80.44; H, 6.16; N,
4.08. Experimental: C, 80.30; H, 6.10; N, 3.91.
Isobutyl pyren-4-ylcarbamate (compound
2)
mp 146–148°C; IR (KBr disc, cm−1): 3282,
3276, 1696, 1560, 1527, 1241, 1112, 971, 865; 1H-NMR δ
(CDCl3, ppm): 1.01 (m, 6H, methyl), 2.03 (m, 1H,
methine), 4.06 (d, 2H, J=6.49 Hz, methylene), 7.21 (broad s,
NH), 7.98–8.14 (m, 9H, Ar-H); 13C-NMR (CDCl3, ppm) δ:
19.23 (2C), 28.14, 71.89, 120.12, 124.87, 124.97(2C), 125.23 (2C),
125.35, 125.42 (2C), 126.21, 126.66, 127.39, 127.88, 130.70,
130.92, 131.49, 155.11. Anal. Calcd. for
C21H19NO2: C, 79.47; H, 6.03; N,
4.41. Experimental: C, 79.31; H, 5.91; N, 4.22.
N-(isobutoxymethyl)pyren-4-amine
(compound 3)
mp 76–78°C; IR (KBr disc, cm−1): 2692,
2669, 2643, 2358, 1600, 1514, 1282, 827; 1H-NMR δ
(CDCl3, ppm): 0.94 (m, 6H, methyl), 2.08 (m, 1H,
methine), 3.01 (m, 2H, methylene), 3.30 (m, 2H, methylene), 4.71
(broad s, NH), 7.31–8.06 (m, 9H, Ar-H); 13C-NMR δ
(CDCl3, ppm): 21.88, 28.17, 31.24, 108.27, 116.53,
119.44, 122.45, 123.03, 123.24, 123.36, 123.87, 124.21, 125.71,
125.84, 126.01, 126.36, 126.54, 126.76, 127.12, 127.93, 143.59.
Anal. Calcd. for C21H21NO: C, 83.13; H, 6.98;
N, 4.62. Experimental: C, 82.89; H, 6.61; N, 4.51.
Spectral analyses of the compounds 1, 2
and 3
Compounds 1, 2 and 3 were dissolved in DMSO at a
concentration of 20 mM. For spectral analyses, compounds were then
diluted to 50 μM in either DMSO or PBS. The absorbance of each
compound, dissolved in either DMSO or PBS, was then analyzed
between the wavelengths of 350–700 nM using a SpextraMaxM5 plate
reader (Molecular Devices, Sunnyvale, CA, USA). In addition,
diluted compounds were excited with light at a wavelength of 350
nM, and the subsequent light emission was analyzed between the
wavelengths of 350–700 nM using a SpextraMaxM5 plate reader.
Mammalian cell culture
HepG2, Hepa1–6, NIH3T3 and HeLa cells were cultured
in DMEM (Invitrogen) containing 10% FBS (Invitrogen), Caco-2 cells
were cultured in DMEM containing 20% FBS and HT-29 cells were
cultured in McCoy's media (Invitrogen) containing 10% FBS. The cell
lines were purchased from American Type Culture Collection (ATCC;
Manassas, VA, USA) and incubated at 37°C with 5%
CO2.
Mammalian cell viability assay
Cells were plated onto a 96-well dish (5,000
cells/well) and incubated overnight at 37°C. The following day,
cells were treated with increasing dosages (3–100 μM) of each
compound, which had been dissolved in DMSO. The DMSO concentration
of treatments was limited to 0.5%, and cells were treated with DMSO
alone (0.5%) or 10 μM doxorubicin as negative and positive controls
for cytotoxicity, respectively. After 48 h, cells were fixed and
cell viability was analyzed using the Sulforhodamine B colorimetric
assay as previously described (30). Absorbance of SRB was measured
utilizing a SpextraMaxM5 plate reader and absorbance values were
normalized to non-treated cells. The normalized cell viability with
increasing drug doses was plotted on a four-parameter logistical
curve, and the IC50 of each compound in each cell line
was calculated using SigmaPlot software (Systat Software, Inc.,
Chicago, IL, USA). Each compound was synthesized in two independent
reactions and used in cell viability assays to generate
dose-response curves. The mean IC50 (in μM), with the
corresponding standard deviation of the two independent synthesis
reactions, was then calculated.
Subcellular localization
Cells (7,200 cells/well) were plated onto Nunc
Lab-TekII 8-chamber slides (Fisher Scientific) and incubated
overnight at 37°C. The following day, cells were treated with
compound (50 μM) for 4 h. After treatment, cells were fixed with
PBS containing 4% formaldehyde for 25 min at room temperature.
Then, they were permeabilized using PBS containing 0.2%
Triton-X-100 for 5 min at room temperature and mounted using
VectaShield mounting medium containing DAPI (Fisher Scientific).
The cells were visualized using a Zeiss AxioImager. Z1
epifluorescent microscope (Carl Zeiss Microimaging, LLC, Thornwood,
NY, USA) with EGFP (Excitation 470/40; Emission 525/50) and DAPI
filters, and images were acquired using AxioVision Rel. 4.6
software (Zeiss).
Results and Discussion
Spectral emission of the compounds 1, 2
and 3
Previous reports have identified that the peak
emission wavelength of pyrene is less than 400 nM, with a shoulder
for that emission peak at approximately 420 nM (31,32).
Similarly, chrysene was previously shown to exhibit a peak emission
wavelength of less than 400 nM, with a shoulder for its emission
peak at approximately 425 nM (33).
Therefore, we aimed to determine whether PAH derivatives 1–3 would
have an impact on the absorption and spectral emission patterns of
these molecules. Although no significant absorbance was detected
for compounds in this study, i.e., compounds 1, 2 and 3, at
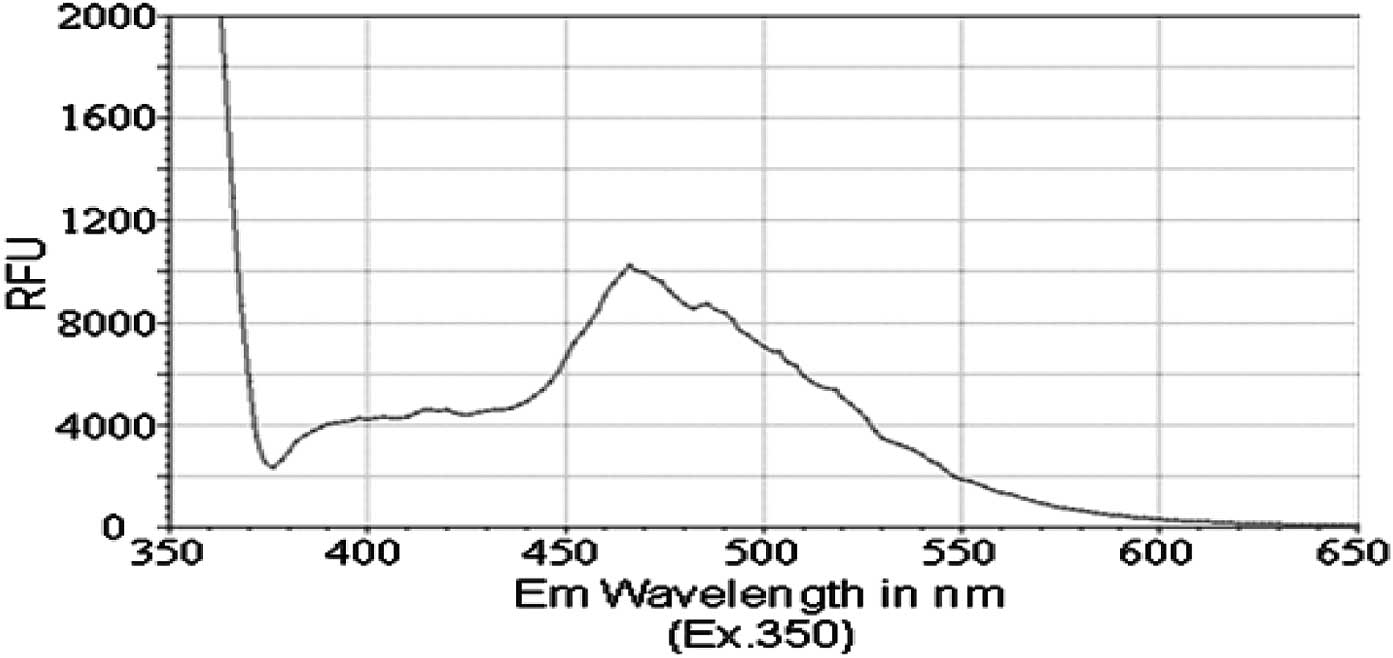
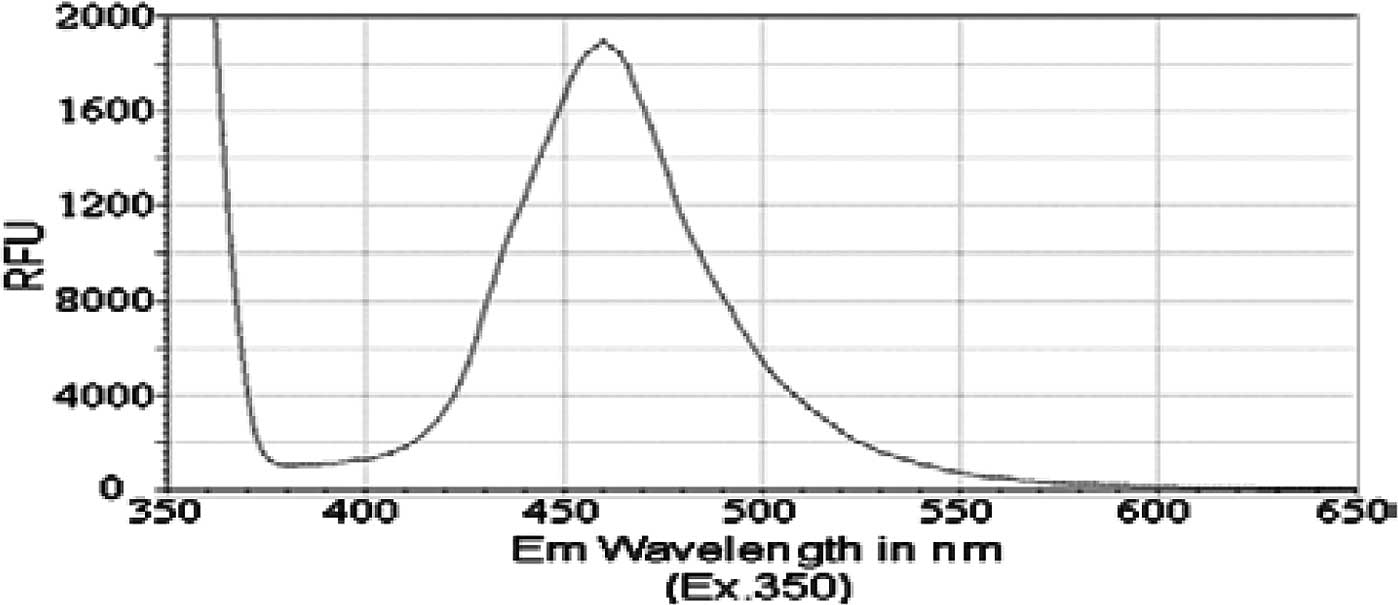
wavelengths between 350 and 650 nM, we detected a fluorescence
emission spectrum for each of the PAH derivatives. The peak
emission wavelength for both isobutyl pyren-4-ylcarbamate (compound
2) and N-(isobutoxymethyl)pyren-4-amine (compound 3) occurred
between 460 and 470 nM (Figs. 2 and
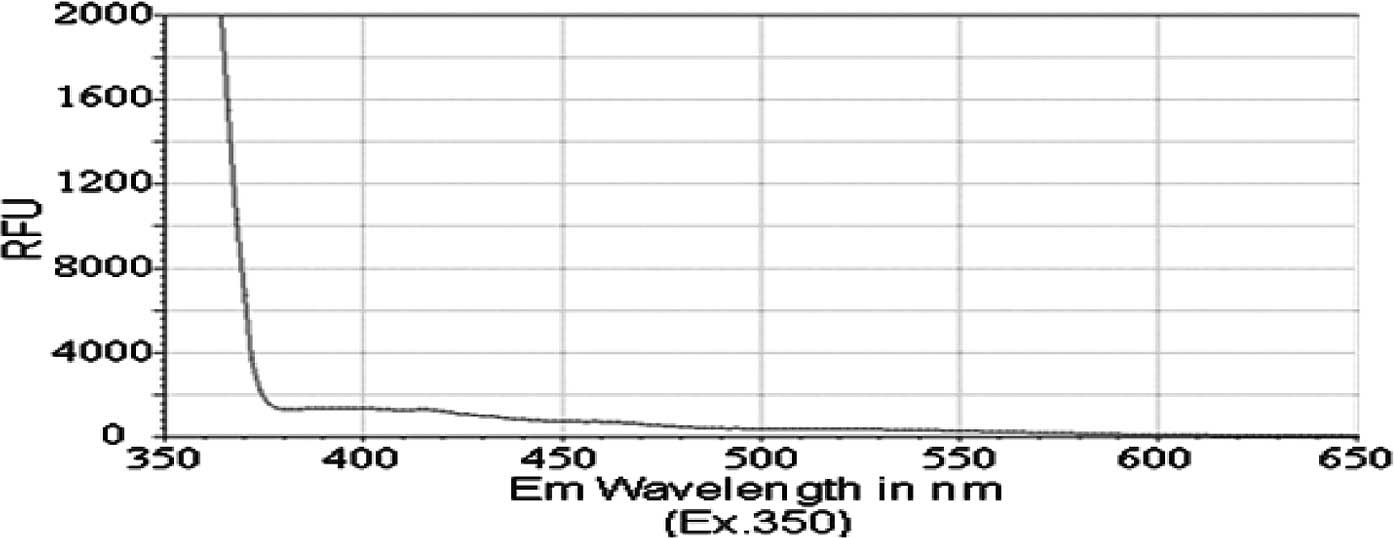
3). However, isobutyl
chrysen-6-ylcarbamate (compound 1) exhibited a much smaller
emission spectra (Fig. 4), with
shoulders for its peak at 420 and 525 nM, when compared to
compounds 2 and 3 (ig. 4 vs. Figs.
1 and 2). The limited
fluorescence of this compound was possibly due to its crystallizing
in aqueous solution, since this emission peak was larger, albeit at
the same wavelength, when compound 3 was dissolved in an organic
solvent, DMSO (data not shown). Nonetheless, these data indicate
that the addition of the long chains to either chrysene or pyrene
caused a red-shift in spectral emission when compared to the PAHs
(chrysene or pyrene).
Decreasing viability of multiple
mammalian cell lines
Previous reports from our group and from other
authors have shown that certain derivatives of PAHs, including
pyrene and chrysene derivatives, reduce the viability of
transformed cell lines (1,20,34),
and some of these PAH derivatives have been reported to reduce cell
viability by induction of apoptosis (34,35).
Therefore, we tested the effects of compounds 1, 2 and 3 on the
viability of a small panel of human and mouse cell lines, including
liver cancer cell lines (HepG2 and Hepa1–6), colon cancer cell
lines (HT-29 and Caco-2), a cervical cancer cell line (HeLa) and
NIH3T3 cells. Colon cancer and the HepG2 liver cancer cell lines
were less susceptible to the effects of the three compounds when
compared to other cell types; Caco-2 cells proved to be
particularly resistant to the effects of these three compounds
(Table I).
 | Table IEstimated IC50 values (in
μM) for the compounds 1, 2 and 3 in a small panel of mammalian cell
lines. |
Table I
Estimated IC50 values (in
μM) for the compounds 1, 2 and 3 in a small panel of mammalian cell
lines.
| Compounds | HepG2 | Hepa1-6 | Caco-2 | HT-29 | HeLa | NIH3T3 |
|---|
| 1 | 31.8±12.6b | 9.1±9.1 | >50 | 30.5±10.0 | 10.5±3.80 | 16.4±5.1 |
| 2 | 30.4±5.0 | 5.9±2.5 | 36.3±3.6 | 20.7±3.8 | 16.2±3.80 | 13.5±11.6 |
| 3 | 39.6±7.1 | 9.9±5.2 | >50 | 14.0±5.7 | 9.8±1.03 | 12.5a |
| Cisplatin | 7.0 | 4.0 | 10.8 | 16.8 | 11.7 | 8.5 |
However, each compound was capable of reducing the
viability of Hepa1–6, HeLa and NIH3T3 cells with an estimated
IC50 value of 16 μM or lower (Table I). Although compound 2 was generally
slightly more cytotoxic than compound 1, little difference in
cytotoxicity was noted when comparing the pyrene (2) and chrysene (1) PAH-coupled compounds. Similarly, little
difference in cytotoxicity was observed when comparing compounds 2
and 3, suggesting that reduction of the carbamate group had little
effect on the pyrene-coupled PAHs.
Since each of the three compounds was capable of
reducing cell viability, at least of certain cancer and non-cancer
cell lines, we also aimed to determine whether this reduction in
cell viability occurred through the induction of apoptosis. We
examined whether compounds 1, 2 and 3 treatment led to increased
caspase 3/7 activity or increased DNA fragmentation (as measured by
a TUNEL assay). We did not detect an increase in either of these
apoptotic features in either HepG2 or Hepa1–6 cells in response to
treatment with any of the three compounds analyzed (data not
shown). Taken together, these data suggest that the ability of
compounds 1, 2 and 3 to reduce cell viability occurs through an
apoptosis-independent mechanism.
A wide variety of planar ring systems are capable of
intercalating with DNA, giving rise to many drugs that possess
chemotherapeutic activity. In this context, three new polyaromatic
derivatives containing polar side chains were systematically
synthesized and investigated. The primary mode of action of these
intercalators is believed to be their reversible binding to nuclear
DNA, which causes inhibition of the replication process and, thus,
cell death. As is well-known, cytotoxicity is not only dependent on
the ability to interact with DNA. Instead, the drug must be capable
of interacting with DNA to form a stable ternary
DNA-intercalator-enzyme complex with a relatively long half-life in
such a way that the enzymatic process cannot progress (11). Therefore, the low level of
cytotoxicity observed for compounds 1–3 may be ascribed to an
inability to access nuclear DNA or a low binding association
(including a low affinity or highly transient inter-action) to
DNA.
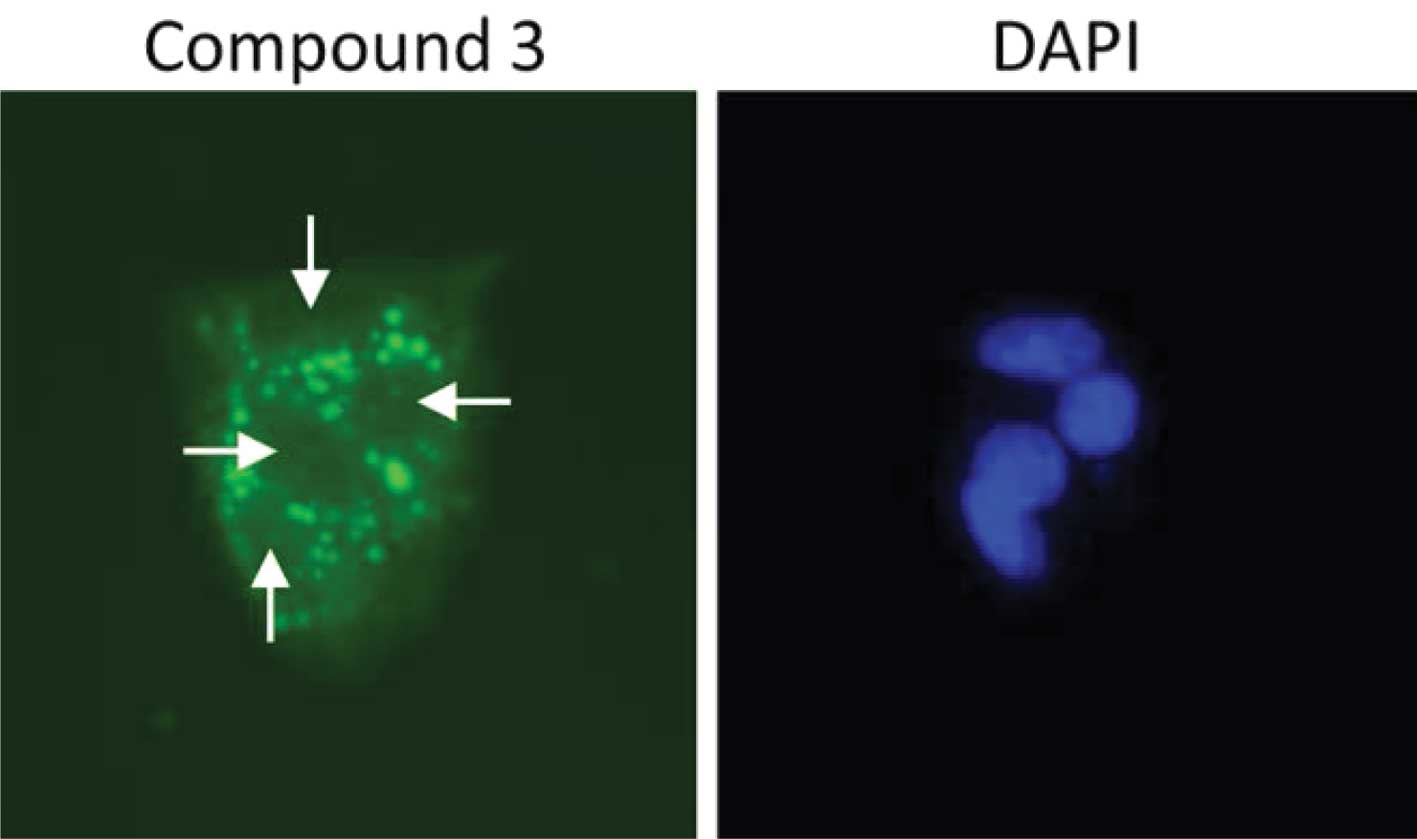
Polyaromatic compounds 1–3 accumulate
outside the cell nucleus
To determine whether the three novel polyaromatic
compounds (i.e., compounds 1–3) were capable of accessing the
nuclear DNA of cells, we examined their auto-fluorescent
properties. HepG2 cells were treated with each compound (50 μM).
Cells were then fixed, stained with DAPI and observed using an
epifluorescent microscope. Each of the three compounds was capable
of being detected using a filter for enhanced green fluorescent
protein, in accordance with their spectral emission properties
(Figs. 2–4). We did not detect any of the three
polyaromatic compounds in cell nuclei under these conditions,
although compounds 1 and 2 formed crystals at these concentrations
(data not shown). Interesetingly, compound 3 accumulated in a
punctate pattern outside the cell nuclei (Fig. 5). This pattern is similar to a
previous study (34) showing that a
pyrene derivate accumulated in the cytosol of cells. These data
suggest that the low level of cytotoxicity observed for these
compounds is more likely due to their inability to access nuclear
DNA in cells rather than a low binding affinity for DNA, although
no studies have been performed to assess the DNA binding affinity
of these compounds.
In conclusion, the present synthetic protocol allows
for the preparation of three new polyaromatic compounds. Structure
elucidation was carried out by various spectroscopic as well as
elemental analyses. The addition of the long-chains to either
chrysene or pyrene caused a red-shift in spectral emission when
compared to the corresponding PAH itself. Furthermore, the
cytotoxicity of the three novel polyaromatic compounds was
evaluated in vitro in a panel of mammalian cell lines.
Results showed that compound 2 exhibited better cytotoxicity
compared to cisplatin against the HeLa cancer cell line, whereas
compound 3, the pyrenyl ether, demonstrated better cytotoxicity
against colon (HT-29) as well as cervical (HeLa) cancer cell lines.
In summary, three new and promising anticancer PAH derivatives have
been synthesized, and structural modification of these compounds is
in progress. In addition, the method for synthesis of compounds 1–3
reported in this study may have applications in other areas of
research.
Acknowledgements
The authors gratefully acknowledge the funding
support from the National Cancer Institute (NIH/NCI-P20, Grant#
5P20CA138022-02).
References
|
1
|
Banik BK and Becker FF: Synthesis,
electrophilic substitution and structure-activity relationship
studies of polycyclic aromatic compounds towards the development of
anticancer agents. Curr Med Chem. 8:1513–1533. 2001. View Article : Google Scholar
|
|
2
|
Lerman LS: Structural considerations in
interaction of DNA and acridines. J Mol Biol. 3:18–30. 1961.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lerman LS: Structure of DNA-acridine
complex. Proc Natl Acad Sci USA. 49:94–102. 1963. View Article : Google Scholar
|
|
4
|
Brana MF, Cacho M, Gradillas A, de
Pascual-Teresa B and Ramos A: Intercalators as anticancer drugs.
Curr Pharm Des. 7:1745–1780. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martinez R and Chacon-Garcia L: The search
of DNA- intercalators as antitumoral drugs: what it worked and what
did not work. Curr Med Chem. 12:127–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Malonne H and Atassi G: DNA topoisomerase
targeting drugs: mechanisms of action and perspectives. Anticancer
Drugs. 8:811–822. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Banik BK and Becker FF: Polycyclic
aromatic compounds as anticancer agents: structure-activity
relationships of chrysene and pyrene derivatives. Bioorg Med Chem.
9:593–605. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wunz TP, Craven MT, Karol MD, Hill GC and
Remers WA: DNA-binding by antitumor anthracene-derivatives. J Med
Chem. 33:1549–1553. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iyengar BS, Dorr RT, Alberts DS, Solyom
AM, Krutzsch M and Remers WA: 1,4-disubstituted anthracene
antitumor agents. J Med Chem. 40:3734–3738. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dorr RT, Liddil JD, Sami SM, Remers W,
Hersh EM and Alberts DS: Preclinical antitumor activity of the
azonafide series of anthracene-based DNA intercalators. Anticancer
Drugs. 12:213–220. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rescifina A, Chiacchio MA, Corsaro A, De
Clercq E, Iannazzo D, Mastino A, Piperno A, Romeo G, Romeo R and
Valveri V: Synthesis and biological activity of isoxazolidinyl
polycyclic aromatic hydrocarbons: potential DNA intercalators. J
Med Chem. 49:709–715. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamal A, Ramesh G, Srinivas O and Ramulu
P: Synthesis and antitumour activity of pyrene-linked pyrrolo
[2,1-c][1,4]benzodiazepine hybrids. Bioorg Med Chem Lett.
14:471–474. 2004.PubMed/NCBI
|
|
13
|
Bair KW, Tuttle RL, Knick VC, Cory M and
McKee DD: (1-Pyrenylmethyl) amino alcohols, a new class of
antitumor DNA intercalators – discovery and initial amine
side-chain structure activity studies. J Med Chem. 33:2385–2393.
1990.PubMed/NCBI
|
|
14
|
Bair KW, Andrews CW, Tuttle RL, Knick VC,
Cory M and McKee DD: 2-[(arylmethyl)amino]-2-methyl-1,3-propanediol
DNA intercalators – an examination of the effects of aromatic ring
variation on antitumor-activity and DNA-binding. J Med Chem.
34:1983–1990. 1991.
|
|
15
|
Demeunynck M, Bailly C and Wilson WD: DNA
and RNA Binders. Wiley-VCH. Weinheim: 2002. View Article : Google Scholar
|
|
16
|
De Vita VT, Hellman S and Rosenberg SA:
Cancer: Principles and Practice of Oncology. 6th edition.
Lippincott Williams and Wilkins; Philadelphia, PA: 2001
|
|
17
|
Lown JW: Anthracycline and anthraquinone
anticancer agents: current status and recent developments.
Pharmacol Ther. 60:185–214. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar
|
|
19
|
Banik BK, Basu MK and Becker FF: Novel
disubstituted chrysene as a potent agent against colon cancer.
Oncol Lett. 1:1033–1035. 2010.PubMed/NCBI
|
|
20
|
Banik BK and Becker FF: Novel
6,12-disubstituted chrysene as potent anticancer agent: synthesis,
in vitro and in vivo study. Eur J Med Chem. 45:4687–4691. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Banik BK, Mukhopadhyay C and Becker FF:
Synthesis and biological evaluation of novel dibenzofluorene
derivatives as anticancer agents. Oncol Lett. 1:309–311. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Banik BK, Samajdar S and Becker FF:
Asymmetric synthesis of anticancer β-lactams via Staudinger
reaction. Mol Med Reports. 3:319–321. 2010.
|
|
23
|
Banik BK and Becker FF: Selective
anticancer activity of β-lactams derived from polyaromatic
compound. Mol Med Reports. 3:315–316. 2010.
|
|
24
|
Banik BK, Banik I and Becker FF:
Asymmetric synthesis of anticancer β-lactams via Staudinger
reaction: utilization of chiral ketene from carbohydrate. Eur J Med
Chem. 45:846–848. 2010.
|
|
25
|
Banik BK, Banik I and Becker FF:
Stereocontrolled synthesis of anticancer β-lactams via the
Staudinger reaction. Biorg Med Chem. 13:3611–3622. 2005.
|
|
26
|
Banik BK, Becker FF and Banik I: Synthesis
of anticancer β-lactams: mechanism of action. Biorg Med Chem.
12:2523–2528. 2004.
|
|
27
|
Banik I, Becker FF and Banik BK:
Stereoselective synthesis of β-lactams with polyaromatic imines:
entry to new and novel anticancer agents. J Med Chem. 46:12–15.
2003.
|
|
28
|
Becker FF, Mukhopadhyay C, Hackfeld L,
Banik I and Banik BK: Polycyclic aromatic compounds as anticancer
agents: synthesis and biological evaluation of dibenzofluorene
derivatives. Bioorg Med Chem. 8:2693–2699. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Becker FF and Banik BK: Polycyclic
aromatic compounds as anticancer agents: synthesis and biological
evaluation of some chrysene derivatives. Biorg Med Chem Lett.
8:2877–2880. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vichai V and Kirtikara K: Sulforhodamine B
colorimetric assay for cytotoxicity screening. Nat Protoc.
1:1112–1116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Búcsiová L, Hrdlovič P and Chmela S:
Spectral characteristics of fluorescence probes based on pyrene in
solution and in polymer matrices. J Photochem Photobiol A Chem.
143:59–68. 2001.
|
|
32
|
Hrdlovič P and Chmela S: Spectral
characteristics of probes based on ionic derivatives of pyrene in
polar polymer matrices. J Photochem Photobiol A Chem. 118:137–142.
1998.
|
|
33
|
Spiro M, Vigny P and Hodgson RM:
Fluorescence spectral studies on the metabolic activation of
chrysene by hamster embryo cells. Carcinogenesis. 3:1491–1493.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ohara K, Smietana M, Restouin A, Mollard
S, Borg J, Collette Y and Vasseur J: Amine-guanidine switch: a
promising approach to improve DNA binding and antiproliferative
activities. J Med Chem. 50:6465–6475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Landis-Piwowar KR, Chen D, Cui QC, Minic
V, Becker FF, Banik BK and Dou QP: Apoptotic-inducing activity of
novel polyaromatic compounds in human leukemic cells. Int J Mol
Med. 17:931–935. 2006.PubMed/NCBI
|