Introduction
Currently, chromosomal instability (CIN) and
microsatellite instability (MSI) are considered to be the principal
driving forces of carcinogenesis (1). In the majority of cases these are
mutually exclusive, but epigenetic gene silencing can act as an
additional factor to either (2).
Thus, driven by chromosomal instability, cancers are thought to
progress when gene functions are compromised by loss of chromosomes
or larger parts thereof, by deletions/insertions of a small number
of nucleotides within microsatellites located in the coding regions
of genes or by methylation of cytosines in or near gene promoters,
leading to loss of gene expression. Each of these carcinogenic
driving forces is reflected in a molecular phenotype of cancers
that can be readily assessed in a molecular pathology laboratory.
Specifically, DNA cytophotometry and/or allelotyping with
polymorphic microsatellite markers is used to test for CIN;
microsatellite analysis of markers particularly susceptible to
instability (usually including the Bethesda markers) (3) is used to address MSI, and
methylation-specific PCR (ideally with quantitative approaches such
as MethyLight) (4) is used to
assess whether promoter methylation is frequent in a cancer (the
CpG island methylator phenotype, CIMP). For colorectal carcinoma,
these molecular features underlie a recently proposed ‘molecular
classification’ (5).
In a previous study (6), we described the application of such
molecular assays to a larger consecutive series of colorectal
carcinomas (n=130). Eleven cases were observed that were not wholly
concordant with our predictions and the current view regarding
carcinogenesis. These tumours were diploid by DNA flow cytometry
and no evidence of allelic imbalance at any of the ‘canonical’ loci
of colorectal carcinoma (5q21, 8p21, 9p21, 17p13 and 18q21) was
observed. Additionally, they were microsatellite-stable and lacked
evidence of CIMP when a MethyLight marker panel was used. We
proposed that this unexpected molecular phenotype may be indicative
of an unusual carcinogenic pathway, and suggested the tentative
designation of ‘X-type’ tumours.
As was discussed in our previous study (6), technical limitations of the analyses
may have influenced the results. Notably, DNA cytophotometry is
prone to omit genomic losses if diploidy is maintained. This
happens in uniparental disomy, a molecular feature researchers are
increasingly becoming aware of (7,8).
Furthermore, small amplifications or deletions in loci not targeted
by our microsatellite marker panel would have escaped detection by
both allelotyping and DNA cytophotometry. Finally, DNA was
extracted from whole tissues in our previous study (6). Although the tumour-stroma ratio had
been controlled for by microscopic examination of scout slides, the
possibility of false-negative identifications of allelic imbalance
remained.
It may be expected that these technical limitations
are overcome when single nucleotide polymorphism (SNP) array
analysis is conducted with tumour tissue separated from the stroma
by laser-capture microdissection. SNP array analysis simultaneously
interrogates the entire genome of a tumour for copy number and
allele status. Additionally, by laser-capture microdissection,
confounding effects of admixed non-tumour DNA are avoided. Using
this approach, as a first objective of this study, we further
investigated the ‘X-type’ colorectal carcinomas and used DNA from
several of our primary colorectal carcinoma cell lines of various
molecular phenotypes for comparison. As a second objective of
methodological interest, this approach enabled a direct comparison
of results from SNP array and microsatellite analyses.
Materials and methods
Prior written informed consent was obtained from all
patients, and all procedures were approved by the Ethics Committee
of the University of Rostock (reference number II HV 43/2004) in
accordance with generally accepted guidelines for the use of human
material.
Tumour tissues and primary colorectal
carcinoma cell lines
Tumour tissue of sufficient quantity was available
for five of the original 11 ‘X-type’ tumours (6); these five cases were used in this
study. Neoplastic glands were separated from the stroma by
laser-capture microdissection using a PALM laser-capture
microdissection device (Carl Zeiss, Göttingen, Germany).
For comparison, we selected nine of our primary
colorectal carcinoma cell lines from early passages. Molecular
typing of these control cases was implemented as described
(6). Clinicopathological and
molecular data of the tumours included in this study are summarized
in Table I.
 | Table ISummary of clinicopathological and
molecular data of the cases included in the study. |
Table I
Summary of clinicopathological and
molecular data of the cases included in the study.
| Tumour ID | Type | Site | Diameter (cm) | TNM | Molecular type | Passage |
|---|
| HROC 18 | ADC | Right colon | 4.0 | G2pT3pN0cM0 | spSTD | 13 |
| HROC 24 | ADC | Right colon | 2.5 | G2pT2pN0cM0 | spMSI | 4 |
| HROC 32 | ADC | Right colon | 5.0 | G2pT4pN2cM0 | spSTD | 7 |
| HROC 39 | ADC | Right colon | 10.0 | G3pT4pN0cM0 | spSTD | 8 |
| HROC 40 | ADC | Left colon | 6.0 | G3pT3pN1cM0 | CIMP | 3 |
| HROC 43 | ADC | Right colon | 5.0 | G3pT3pN2cM0 | spSTD | 4 |
| HROC 46 | ADC | Right colon | 6.0 | G3pT3pN0cM1 | spSTD | 5 |
| HROC 60 | ADC | Right colon | 3.2 | G2pT2pN0cM0 | CIMP | 6 |
| HROC 69 | ADC | Right colon | 12.0 | G3pT3pN0cM0 | spSTD | 6 |
| T37 | ADC | Left colon | 1.5 | G1pT1(sm3)pN0cM0 | ‘X-type’ | NA |
| T53 | mucCa | Right colon | 12.0 | G1pT3pN0cM0 | ‘X-type’ | NA |
| T97 | ADC | Rectum | 5.5 | G2pT3pN0cM0 | ‘X-type’ | NA |
| T104 | ADC | Right colon | 8.0 | G2pT3pN0cM0 | ‘X-type’ | NA |
| T109 | ADC | Rectum | 5.5 | G2pT2pN1cM0 | ‘X-type’ | NA |
Patients’ normal mucosa (for ‘X-type’ tumours) or
B-lymphocytes (for cell lines) were used to obtain individually
matched non-tumour DNA.
DNA extraction and SNP array
hybridisation
Laser-capture microdissected sample material was
incubated with 8 μl proteinase K (20 mg/ml) overnight at 56°C. DNA
was subsequently purified using the column-based NucleoSpin Tissue
XS kit (Macherey-Nagel, Düren, Germany), according to the
manufacturer’s protocol for microdissected material.
The DNA samples were treated as described in the
Affymetrix Cytogenetics Copy Number Assay User Guide; 500 ng
genomic DNA sample was portioned into two aliquots of 250 ng and
these were cleaved by restriction endonucleases (StyI and
NspI). Following adapter ligation, a reduction of the
genomic complexity was performed by limited cycle preparative PCR.
In alteration to the protocol, the PCR products were cleaned up by
an ultrafiltration procedure using NucleoFast 96 PCR Plates
(Macherey-Nagel). Fragmentation by DNaseI and end labelling was
conducted using the Genome-Wide Human Nsp/Sty 5.0/6.0 Assay kit
(Affymetrix, Santa Clara, CA, USA). The hybridisation of
Genome-Wide Human SNP 6.0 arrays was followed by 16–17 hours
incubation at 50°C in the GeneChip Hybridization Oven 640. After
washing, staining and antibody amplification using the Fluidics
Station 450, the SNP 6.0 arrays were scanned with the Affymetrix
GeneChip Scanner 3000 (7G).
Processing of SNP array hybridisation
data and evaluation
For copy number and loss of heterozygosity (LOH)
analysis, the intensity data files obtained by scanning of the
processed microarrays (CEL files), were imported into the
Genotyping Console Software (Affymetrix). Data processing followed
the implemented standard workflow for SNP 6.0 array and unpaired
analysis was performed using an implemented HapMap sample set of
270 individuals as a reference.
For visualisation, the result files were loaded into
the Affymetrix GTC browser software to display the log2 ratio, copy
number state and LOH state over a RefSeq track. The displays of
tumour DNA hybridisations were directly compared with SNP array
hybridisations of patients’ normal DNA, chromosome by chromosome in
each case. Aberrations of ploidy status and allele status were
recorded under the following categories: i) loss or amplification
of whole chromosomes or chromosome arms; ii) uniparental disomy or
uniparental polysomy of whole chromosomes or chromosome arms; iii)
subchromosomal deletions or amplifications, the chromosomal sites
of which were recorded by reference to the chromosome bar at the
bottom of the viewer, and the cut-off between extensive
deletion/amplification versus microdeletions/microamplifications
(see below) was arbitrarily set to 1 MB of DNA; iv) subchromosomal
uniparental disomy and v) microdeletions or
microamplifications.
Microsatellite analyses
The microsatellite markers employed for allelotyping
targeted the ‘canonical’ colorectal carcinoma loci 5q21, 8p21,
9p21, 17p13 and 18q21; technical details of these assays are
described in our previous study (6). Two dinucleotide markers located at
3p14.2 (D3S1234, D3S1300) were added in the present study. For the
control cases, the majority of microsatellite analyses were
conducted using DNA from the cell lines; only in a minority of
cases was DNA from corresponding xenografts used (details of the
xenografting procedures have been published previously) (9). Microsatellite instability was tested
with the Bethesda markers.
Results
Genomic aberrations recorded by SNP array
analysis
SNP array hybridisations were performed successfully
with genomic DNA extracted from the primary colorectal carcinoma
cell lines and with DNA from laser-capture-microdissected
neoplastic glands of the colorectal carcinoma surgical specimens.
All tumours harboured certain genomic aberrations. However, the
frequency and type of aberration varied between cases.
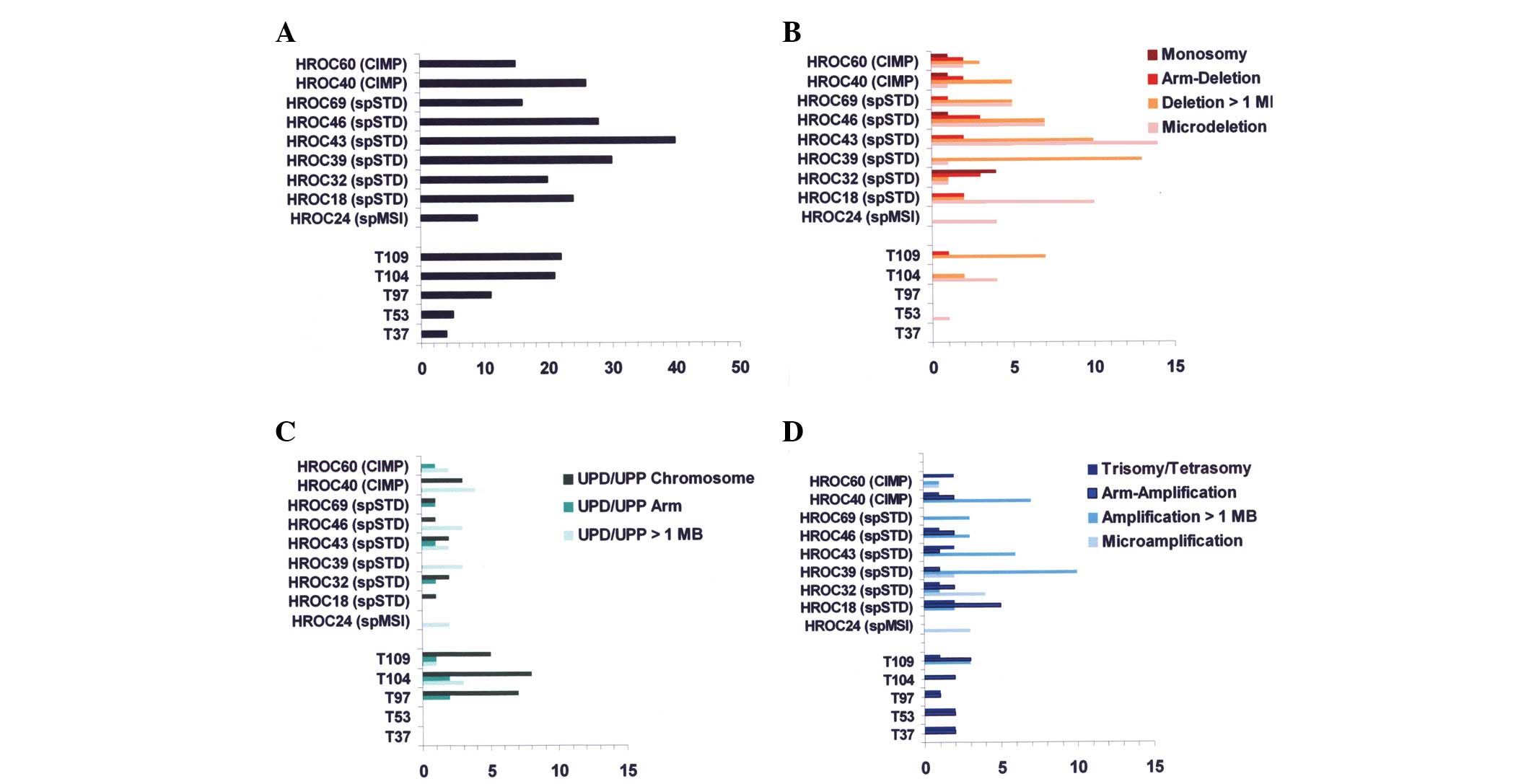
Fig. 1A reveals the
frequencies of all aberration types; very low total numbers were
recorded for two of the ‘X-type’ tumours (T37 and T53, totals of
five and four, respectively) and for HROC 24, which is of the
sporadic MSI-type (a total of nine aberrations). The remaining
tumours, including the three ‘X-type’ tumours T97, T104 and T109,
exhibited higher totals (range, 11–40).
In Fig. 1B–D
frequencies of different types of genomic aberrations are plotted
per case. Fig. 1B reveals that
deletions were numerous in the sporadic standard (spSTD)- and
CIMP-type tumours (range, 8–26). Conversely, T97, T104 and T109
were observed to have uniparental disomies (UPD)/uniparental
polysomies relatively frequently (range, 7–13). However, neither
UPD nor uniparental polysomies were completely absent from the
spSTD-/CIMP-type tumours, nor were deletions from the ‘X-type’
tumours. Notably, UPD/uniparental polysomies in the ‘X-type’
tumours T97, T104 and T109 most often affected whole chromosomes.
Genomic aberrations in the other two ‘X-type’ tumours, T37 and T53,
were trisomies or arm amplifcations (trisomies 13 and 16, and
8p-/20q arm amplifications for T37; trisomies 13 and 20, and
1q-/19q arm amplifications for T53).
All colorectal carcinoma cell lines had at least one
microdeletion (range, 1–14) as did two of the ‘X-type’ tumours (T53
and T104; 1 and 4, respectively). Microdeletions were observed in
the Affymetrix Genotyping Console Browser image as ‘punched out’
losses of hybridisation signals representing up to 1 MB of DNA (but
usually considerably less). Recurrent microdeletions were observed
at 3p14.2 (5 of the colorectal carcinoma cell lines), 20p12.1 (4 of
the colorectal carcinoma cell lines) and 16p13.2 (4 of the
colorectal carcinoma cell lines and 2 of the ‘X-type’ tumours). The
genes belonging to these loci were identified as FHIT, MACROD2 and
A2BP1, respectively. The other microdeletions were distributed over
the genome without any recognisable pattern.
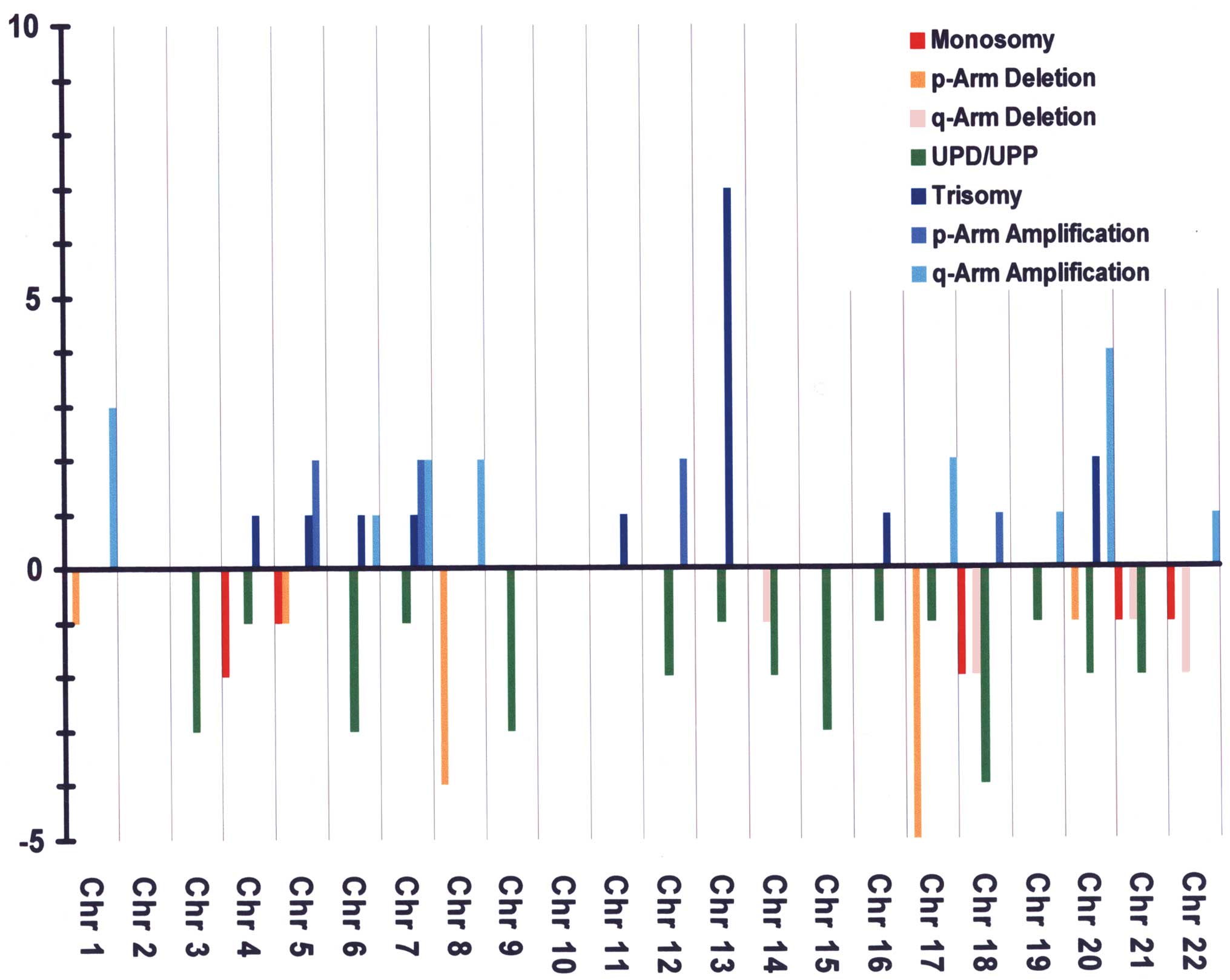
In Fig. 2, the
frequencies and types of genomic aberrations are plotted in
relation to the different chromosomes. As can be observed from this
figure, aberrations were most frequent in chromosomes 17, 18 and
20.
SNP array analysis compared to
allelotyping by microsatellite analysis
A comparative analysis was carried out with DNA from
the nine colorectal carcinoma cell lines and/or xenografts, using
15 polymorphic microsatellite markers that represented 12 loci (as
detailed in Materials and methods). Informative results were
obtained in 100 of the 135 PCR reactions. Allelotyping was not
possible due to the homozygosity of the microsatellite markers in
26 reactions, or not evaluable due to MSI in nine reactions (7
markers for HROC 24, which is of the spMSI-type, and 1 marker each
for HROC 18 and HROC 69). Allelotyping showed loss of
heterozygosity (LOH)/allelic imbalance in 61 reactions. In all but
one case, this was seen to involve complete loss of the allele in
the electropherograms; LOH in its strict sense. As shown in
Table II, there was overall
concordance between the two methods, with discrepancies recorded
for seven of the 100 analyses (7.0%). Deletion or uniparental
disomy, recorded by SNP array analysis, without evidence of allelic
loss/allelic imbalance by microsatellite analysis was observed
twice and once, respectively. Allelic loss or allelic imbalance,
identified by microsatellite analysis, without evidence of genomic
aberrations in the SNP arrays was recorded four times.
| Table IIComparison of results from
microsatellite (MS) and single nucleotide polymorphism (SNP) array
analyses using DNA from colorectal carcinoma cell lines. |
Table II
Comparison of results from
microsatellite (MS) and single nucleotide polymorphism (SNP) array
analyses using DNA from colorectal carcinoma cell lines.
| MS complete loss | MS incomplete
loss | MS no loss |
|---|
| SNP deletion | 37 | - | 2 |
| SNP UPD | 20 | - | 1 |
| SNP no
aberration | 3 | 1 | 36 |
In our previous publication, ‘X-type’ tumours were
defined by flow-cytometric diploidy, absence of MSI and absence of
CIMP, in addition to absence of allelic imbalance at the canonical
loci 5q21, 8p21, 9p21, 17p13 and 18q21. These analyses had been
conducted using DNA from whole tissue cryostat sections with tumour
fractions greater than 50% (6).
Unexpectedly, for three of the tumours classified as ‘X-type’, the
SNP arrays revealed UPD or deletion in nine of these loci.
Therefore, allelotyping was then performed with DNA from
laser-capture microdissections, as used for the SNP arrays. This
showed allelic losses concordant with the SNP arrays in seven of
the nine loci (details in Table
III).
| Table IIIResults of single nucleotide
polymorphism (SNP) array and microsatellite (MS) analyses with DNA
extracted from ‘X-type’ tumours after laser-capture
microdissections. |
Table III
Results of single nucleotide
polymorphism (SNP) array and microsatellite (MS) analyses with DNA
extracted from ‘X-type’ tumours after laser-capture
microdissections.
| ID | 5q21
| 8p21
| 9p21
| 17p13
| 18q21
|
|---|
| SNP | MS | SNP | MS | SNP | MS | SNP | MS | SNP | MS |
|---|
| T37 | - | - | - | - | - | - | - | - | - | - |
| T53 | - | - | - | - | - | - | - | - | - | - |
| T97 | - | - | - | - | UPD | LOH | DEL | LOH | UPD | LOH |
| T104 | UPD | - | - | - | UPD | LOH | UPD | LOH | UPD | LOH |
| T109 | - | - | - | - | - | - | DEL | - | UPD | LOH |
Discussion
The primary objective of this study was to further
investigate the provisional molecular ‘X-type’ of colorectal
carcinoma. Specifically, we addressed whether there would be
evidence for, or at least an indication of, the regulating forces
of carcinogenesis and tumour progression that differed from the
orthodox triad of CIN, MSI and CIMP. Using SNP array analyses, we
searched the entire genome for copy number changes and allelic
losses, avoiding the potentially contaminating effects of
non-neoplastic tissue by laser-capture microdissection. At present,
only one study concerning colorectal carcinoma has been published
that uses this approach (8).
Two of the ‘X-type’ tumours included in this study
(T37 and T53) were markedly different from the rest of the cases;
they differed from the remaining three ‘X-type’ tumours as well as
from the control cases (Fig. 1).
Apart from a single microdeletion observed in T53, these tumours
did not have any allelic losses, neither by deletions nor by UPD.
Thus, these two tumours appeared to be as similar as possible to an
unorthodox molecular phenotype of colorectal carcinoma. Considering
that they were selected from 130 colorectal carcinomas originally,
this appears to be an exceedingly rare molecular phenotype.
However, what makes these tumours unique and whether this molecular
phenotype could have any pathogenetic implications are currently
unknown. Notably, these colorectal carcinomas are at the borders of
the usual clinicopathological spectrum, but in different ways to
each other. T53 is a well-differentiated mucinous carcinoma without
regional or distant metastases, but is well-advanced locally
(diameter 12.0 cm; pT3). This relatively unusual histotype may be
the explanation for the unusual molecular phenotype. Conversely,
T37 is a small cancer (diameter 2.5 cm; pT1, though infiltrating
well into the submucosa, sm3; node-negative), but is otherwise a
morphologically non-descript adenocarcinoma with a moderate degree
of tumour budding and with nuclear β-catenin translocation by
immunohistochemistry (10). An
explanation may be that this ‘early’ cancer simply did not have
enough time to develop the load of genomic aberrations observed in
the other tumours. The explanation for T37 may thus initially
appear trivial; however, certain implications are evident. For
example, it may be argued that if an ‘early’ colorectal carcinoma
such as T37 is capable of sharing the phenotype of invasion with
any other type of colorectal carcinoma (including metastasizing
tumours) but does not share their molecular phenotype, then the
function of the orthodox carcinogenic triad of CIN, MSI and CIMP
may be a late effector in tumour progression, particularly in
metastasizing disease. However, if invasiveness may be acquired
without CIN, MSI or CIMP, and the pathogenetic function of this
molecular phenotype is in tumour progression (the metastasizing
course of the disease), then the relatively heavy loads of genomic
loss observed in a number of our colorectal carcinomas without
metastasis indicate that many of these may be functionally
irrelevant. Such ‘background noise’ of genomic changes is a
well-recognised phenomenon (11),
but generalising from our observations, its extent and relevance in
the interpretation of molecular analyses may be significantly
underestimated. Moreover, it may transpire that it is extremely
difficult to overcome this background noise in molecular studies,
since for practical and ethical reasons, small cancers are
significantly under-represented in tumour specimen collections,
including in our own tumour bank.
However, for the remaining three tumours classified
as ‘X-type’ (T97, T104 and T109), repeated microsatellite analyses
with DNA from laser-capture microdissected tumour tissue was
prompted by the SNP array analyses that had revealed UPD or
deletions at certain loci tested in our previous study (Table III). These repeated microsatellite
analyses revealed that LOH was indeed present at these loci in the
majority of cases, having gone undetected in the initial tests with
DNA from whole tumour tissues. Subsequently, these three tumours
were reclassified as spSTD-type colorectal carcinoma, although UPD
was a relatively frequent molecular feature of them and may explain
their diploid status by DNA flow cytometry.
Our initial failure to detect allelic imbalance when
allelotyping these three tumours raises an important methodological
issue. The overwhelming majority of published LOH studies rely on
DNA from whole tumour tissues. Typically, such as in our initial
study, the tumour content is assessed by microscopic examination of
histological sections, taken as sufficient at a fraction of 50–80%
and LOH is scored if the tumour-normal ratios are below or above
the arbitrary limits of 0.5 or 2.0, respectively. As noted
previously (12), this
microsatellite analysis of whole tissue DNA in reality is not a
study of LOH but of allelic imbalance. Therefore, allelotyping is
the correct designation for this procedure. As demonstrated in the
present study, allelotyping carries a significant chance of
false-negative determinations. Furthermore, as amplifications are
not detected by this method, allelotyping is also prone to
false-positive determinations. These methodical drawbacks strongly
detract from the value of the majority of LOH studies of colorectal
carcinomas or other solid tumours. LOH sensu strictu can
only be diagnosed if, as was performed in this study, either tumour
cell lines/xenografts or tumour tissue from laser-capture
microdissections are used for the microsatellite analyses, and if
complete absence of one allele is then observed.
Notably, in the majority of cases, microsatellite
analyses at the loci tested in this study revealed a complete loss
of one allele (Table II); an
incomplete loss was recorded in only one instance. Thus, although
deletion events or UPD may very often be background noise as has
been discussed previously, they also have the potential to
compromise gene function as proposed in the suppressor pathway
concept. Therefore, when assessing the functional role of LOH by
deletions or UPD in a given case, how these combine with mutations
in the remaining alleles should be investigated. It has been
demonstrated in colorectal carcinoma that gene mutations
preferentially target a selection of a number of candidate cancer
(CAN) genes, which are typically members of a signal transduction
pathway that thereby undergoes dysregulation (13). The challenging task for researchers
to negotiate is to find the relevant gene mutations by whole genome
sequencing procedures, and then to compare them with the
genome-wide allele status that can only be determined by SNP array
analysis. In this context, the secondary objective of our study may
be of methodological interest as follows.
As a secondary objective, the low-passage colorectal
carcinoma cell lines of various molecular phenotypes that were
included in this study allowed us to address how well LOH (by
deletion or UPD) is represented in the SNP array analyses as
compared with microsatellite analysis. If microsatellite analysis
is informative and reveals a complete loss of one allele at a given
locus, it may be regarded as a standard to compare with. Overall,
discrepancies between microsatellite and SNP array analyses were
observed in 7% of tests. Assuming microsatellite analysis is the
standard, false-negative and false-positive determinations for LOH
by SNP array analysis were observed in 4 and 3% of tests,
respectively. We consider this to be a relatively low rate,
attesting to the proficiency of the SNP array technique. However,
it should be considered when interpreting data. To our knowledge,
such comparisons have not been published and this may therefore be
of interest for researchers who apply these techniques.
Microdeletions were a noteworthy observation in the
SNP arrays. While the majority were distributed over the genome in
no apparent order, recurrence was observed at three gene loci,
viz., FHIT, MACROD2 and A2BP1, introducing the possibility of a
potential functional role. In previous SNP array studies of
colorectal carcinoma, deletions at 16p13.2 centering on A2BP1 have
been described in a single study by Andersen et al(14). These authors also sequenced the gene
in cases with deletions, failing to find mutations, and the
deletions did not appear to correlate with differences of gene
expression. Similiarly, correlative microsatellite/expression
studies of FHIT did not reveal an effect of LOH on gene expression
(15). Aberrations of the MACROD2
gene have not previously been identified for colorectal carcinoma;
studies have only considered MACROD2 deletions in population-based
genotyping (16). Therefore, though
counterintuitive, there is no evidence to suggest that the ‘punched
out’ gene losses so indicative of a functional role are implicated
in the suppressor pathway of colorectal carcinoma. Alternatively,
the recurrent loss of genetic material may arise as a consequence
of recurrent translocation events that, as has been appreciated
recently, are not as rare in solid tumours as previously thought
(17). Notably, Andersen et
al’s study of interphase and metaphase-FISH for 16p13.2 using
four commercial colorectal carcinoma cell lines for SW620 revealed
a balanced translocation (t[3;16]) with the breakpoint centering on
16p13; for the other three cell lines, however, the deletions were
interstitial deletions (14).
Furthermore, rearrangements of the MACROD2, A2BP1 and FHIT genes in
colorectal carcinoma were observed in a study by Bass et
al(18), putatively due to
structural fragility.
Taken together, in this study we have demonstrated
that colorectal carcinomas may develop without the classic
molecular features of CIN, MSI and/or CIMP, but this is a rare
event. We observed that UPD is frequent in the context of CIN and
most likely does not define a separate molecular phenotype.
Furthermore, with regard to methodology, our study supports the
notion that SNP array hybridisations are rather reliable for
genome-wide detection of deletions and UPD, but strongly
discourages LOH analyses with polymorphic microsatellite markers
for DNA from whole tissues.
Acknowledgements
This study was in part supported by a
grant (no. 108919) from the Deutsche Krebshilfe e.V.
References
|
1
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JP: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boland CR, Thibodeau SN, Hamilton SR,
Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA,
Fodde R, Ranzani GN and Srivastava S: A National Cancer Institute
workshop on microsatellite instability for cancer detection and
familial predisposition: development of international criteria for
the determination of microsatellite instability in colorectal
cancer. Cancer Res. 58:5248–5257. 1998.
|
|
4
|
Ogino S, Cantor M, Kawasaki T, Brahmandam
M, Kirkner GJ, Weisenberger DJ, Campan M, Laird PW, Loda M and
Fuchs CS: CpG island methylator phenotype (CIMP) of colorectal
cancer is best characterised by quantitative DNA methylation
analysis and prospective cohort studies. Gut. 55:1000–1006. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jass JR: Classification of colorectal
cancer based on correlation of clinical, morphological and
molecular features. Histopathology. 50:113–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ostwald C, Linnebacher M, Weirich V and
Prall F: Chromosomally and microsatellite stable colorectal
carcinomas without the CpG island methylator phenotype in a
molecular classification. Int J Oncol. 35:321–327. 2009.PubMed/NCBI
|
|
7
|
Gaasenbeek M, Howarth K, Rowan AJ, Gorman
PA, Jones A, Chaplin T, Liu Y, Bicknell D, Davison EJ, Fiegler H,
Carter NP, et al: Combined array-comparative genomic hybridization
and single-nucleotide polymorphism-loss of heterozygosity analysis
reveals complex changes and multiple forms of chromosomal
instability in colorectal cancers. Cancer Res. 66:3471–3479. 2006.
View Article : Google Scholar
|
|
8
|
Andersen CL, Wiuf C, Kruhøffer M,
Korsgaard M, Laurberg S and Ørntoft TF: Frequent occurrence of
uniparental disomy in colorectal cancer. Carcinogenesis. 28:38–48.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Linnebacher M, Maletzki C, Ostwald C,
Klier U, Krohn M, Klar E and Prall F: Cryopreservation of human
colorectal carcinomas prior to xenografting. BMC Cancer.
10:362–371. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prall F, Weirich V and Ostwald C:
Phenotypes of invasion in sporadic colorectal carcinomas related to
aberrations of the adenomatous polyposis coli (APC) gene.
Histopathology. 50:318–330. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tomlison IPM, Lambros MBK and Roylance RR:
Loss of heterozygosity analysis: Practically and conceptually
flawed? Genes Chromosomes Cancer. 34:349–353. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Devilee P, Cleton-Jansen AM and Cornelisse
CJ: Ever since Knudson. Trends in Genet. 17:569–573. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wood LD, Parsons DW, Jones S, Lin J,
Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N,
et al: The genomic landscapes of human colorectal breast and
colorectal cancers. Science. 318:1108–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Andersen CL, Lamy P, Thorsen K, Kjeldsen
E, Wikman F, Villesen P, Oster B, Laurberg S and Orntoft TF:
Frequent genomic loss at chr16p13.2 is associated with poor
prognosis in colorectal cancer. Int J Cancer. 129:1848–1858. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wierzbicki PM, Adrych K, Kartanowicz D,
Dobrowolski S, Stanislawowski M, Chybicki J, Godlewski J,
Korybalski B, Smoczynski M and Kmiec Z: Fragile histidine triad
(FHIT) gene is overexpressed in colorectal cancer. J Physiol
Pharmacol. S4:63–70. 2009.PubMed/NCBI
|
|
16
|
Bradley WE, Raelson JV, Dubois DY, Godin
E, Fournier H, Privé C, Allard R, Pinchuk V, Lapalme M, Paulussen
RJ and Belouchi A: Hotspots of large rare deletions in the human
genome. PloS One. 5:e94012010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mitelman F, Johansson B and Mertens F: The
impact of translocations and gene fusions on cancer causation.
Nature Rev Cancer. 7:233–245. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bass AJ, Lawrence MS, Brace LE, Ramos AH,
Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A,
Jing R, et al: Genomic sequencing of colorectal adenocarcinomas
identifies a recurrent VTI1A-TCF7L2 fusion. Nature Genetics.
43:964–968. 2011. View
Article : Google Scholar : PubMed/NCBI
|