Introduction
Human colorectal cancer (CRC) affects 6% of the
world’s population and is the third most common cancer in Western
countries (1). If metastasis
occurs, the 5-year survival rate of CRC is ∼10% (2). CRC in Korea was known to be less
prevalent than in Western countries; however, the 2011 cancer
survey by the Korean Society of coloproctology reported that the
incidences of CRC in Korean males and females were 46.9 and 25.6
per 100 thousand people, respectively, and the incidence is
expected to increase up to two-fold by 2030. The upsurge of CRC
incidence in Korea may be related to changes in lifestyle, such as
the Western culinary culture. CRC is now becoming a serious public
health problem.
Uncontrolled proliferation of cancer cells is
closely associated with genetic mutations in cell cycle regulators,
signal-regulated kinases and tumor suppressors. Therefore, the
majority of the strategies for developing chemotherapeutic
anticancer agents target these regulatory proteins to inhibit tumor
growth. In CRC, genetic mutations in Apc, KRAS, PIK3CA, p53
and Src are known to promote tumor growth (3). In addition to genetic mutations,
environmental factors, such as obesity, are risk factors for CRC.
The effectiveness of chemotherapy against CRC varies among patients
due to the heterogeneous nature of CRC; hence, a specific
therapeutic regime may be required for each individual CRC case
(3).
Medicinal herbs used in traditional oriental
medicine are attractive sources for developing novel therapeutics
or preventives, since they have been used for thousands of years in
clinics. Therefore, they have been pre-validated for effectiveness
and are expected to have fewer safety issues than chemically
synthesized drugs. Asiasarum heterotropoides var.
mandshuricum F. Maekawa is a perennial plant that grows in East
Asian countries, including Korea, China and Japan. The radix of
Asiasarum heterotropoides var. mandshuricum F. Maekawa
(A. radix) is called seshin in Korean, saishin
in Japanese, or Chinese wild ginger in English. In traditional
Korean medicine, A. radix has been used as an antibiotic or
anti-nociceptive for treating arthralgia and diverse pains
(4). In Japanese herbal medicine
(kampo), A. radix has been used as an antitussive and
anti-allergy drug (5). The majority
of research on A. radix focuses on its anti-allergy or
anti-inflammatory effects. The aqueous extract of A. radix
was shown to inhibit formalin-induced hyperalgesia via
N-methyl-D-aspartate (NMDA) receptors (5) and to have an anti-allergy potential
through inhibition of IgE production in in vitro and in
vivo models (6). Hashimoto
et al reported that several single compounds derived from
the methanol extract of A. radix presented anti-allergy
effectiveness in in vitro passive cutaneous anaphylaxis
(PCA) tests and inhibited the activity of 5-lipoxygenase in rat
basophilic leukemia (RBL-1) cells (7). As a neuropharmaceutical agent, the
methanol extract of A. radix exerted significant inhibitory
effects on α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
(AMPA)-induced excitotoxicity in PC12 rat neuroblastoma cells
(8). In addition, the methanol
extract and its subfractions enhance memory in rats through the
activation of the insulin receptor and the extracellular signal
regulated kinase (ERK1/2) and through the inhibition of
cholinesterase activities in the hippocampus (9). However, only limited information on
the anticancer effects of A. radix is currently available.
Takasaki et al reported that two hydrophobic lignans
isolated from A. radix, asarinin and xanthoxylol,
demonstrated antitumor-promoting activities on a two-stage
carcinogenesis test using mouse skin and pulmonary tumors (10). However, they did not suggest a
mechanism for the antitumor activity of the two lignans. The aim of
our present study was to evaluate the ethanol extract of
Asiasari radix (EEAR) on the proliferation of human HCT-116
CRC cell lines and to investigate the underlying mechanism of its
anticancer effect.
Materials and methods
Preparation of EEAR
A. radix was purchased from Kwangmyungdang
Medicinal Herbs (Ulsan, Korea). The identification of the
Asiasarum heterotropoides var. mandshuricum F. Maekawa was
confirmed by Dr Go Ya Choi, Herbal Medicine Research Division,
Korea Institute of Oriental Medicine. A voucher specimen
(KIOM-CRC-2) was deposited at the Cancer Research Center (KM-Based
Herbal Drug Research Group, Herbal Medicine Research Division).
Dried A. radix (200 g) was finely ground and immersed in 70%
(v/v) ethanol (100 g/l). The solvent extraction was carried out by
two consecutive ultrasonications for 1 h. The extracts were
filtered through Whatman No. 2 filter paper and the excess solvent
was evaporated under reduced pressure using a rotary evaporator
(EYELA, Japan) at 40°C. The powdered extract (EEAR, 15.82 g) was
homogenized using a mortar and stored at 4°C for further studies.
The yield of the extract was approximately 17.91% (w/w). A stock
solution of EEAR was prepared at a concentration of 20 mg/ml in
dimethylsulphoxide (DMSO) and stored at −70°C until use.
Cell line and culture condition
Among several human CRC cell lines, HCT-116 is a
favorable in vitro CRC model for investigating the cell
signaling pathways targeted by anticancer chemotherapy (1). HCT-116 cells were obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA) and
grown in McCoy’s 5A medium (Invitrogen Life Technologies, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (Invitrogen), 100
U/ml penicillin and 100 μg/ml streptomycin (Invitrogen) in a
humidified atmosphere of 5% CO2 at 37°C.
Cell proliferation assay
Viable cells were quantified using the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay (Promega, Madison, WI, USA). In brief, cells were
seeded at 5×103 cells/well in a 96-well culture plate
containing 100 μl complete medium 1 day before drug treatment.
Cells were treated with the indicated concentrations of EEAR for 48
h. The DMSO was used as a negative control. The cultured media were
removed and the cells were washed with phosphate-buffered saline
(PBS) before adding the MTS working solution to minimize the
interference of the drugs within the MTS reaction. One hundred
microliters of 20% (v/v) MTS diluted in fresh culture medium was
added and the cells were incubated for a further 30 min at 37°C.
Color development was monitored using a microplate reader at 590 nm
(Molecular Devices, Sunnyvale, CA, USA).
Quantification of apoptosis
The degree of cellular apoptosis was measured using
a commercially available enzyme-linked immunosorbent assay (ELISA)
kit (Cell death detection ELISAPLUS, Roche, Mannheim,
Germany) according to the manufacturer’s instructions. Apoptosis
can be quantified since the amount of mono- and oligonucleosomes
increase in the cytoplasm before plasma membrane breakdown due to
the activation of endogenous endonucleases in apoptotic cells. The
fragmented nucleosomes released into the cytoplasm were captured
and quantified by a sandwich ELISA. In brief, exponentially growing
cells were trypsinized and resuspended in the culture medium. Five
thousand cells in 100 μl culture medium were plated in each well of
a 96-well culture plate and were left to attach to the plate for 24
h before drug treatment. Cells were treated with various
concentrations of EEAR for 24 h and then cell lysates were prepared
using 200 μl of the lysis buffer included in the kit. Twenty
microliters of each sample was subjected to the ELISA assay. The
specific enrichment factors of the mono- and oligonucleosomes in
the cytoplasm (the degree of apoptosis) were calculated using the
following equation: enrichment factor = mU of the sample
(dying/dead cells) / mU of the corresponding negative control
(cells treated with vehicle), where mU = absorbance
(10−3).
Western blot analysis
Changes in intracellular protein levels by EEAR
treatment were determined by western blot analysis. Cells were
seeded at 1×106 cells/dish in 60-mm culture dishes 1 day
before drug treatment. Cells were treated with various
concentrations of EEAR for 24 h. The total protein was extracted
with ice-cold RIPA cell lysis buffer (Thermo Scientific, Rockford,
IL, USA) and the protein concentration was quantified by the
bicinchoninic acid (BCA) colorimetric method (Thermo Scientific).
Equal amounts of proteins were separated on sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and
transferred to nitrocellulose membranes. The protein-blotted
membrane was blocked with a 5% (w/v) skimmed milk solution in 0.1%
(v/v) Tween-20 PBS for 1 h at room temperature (RT) and then probed
with primary antibodies at 4°C overnight. The primary antibodies
were captured with suitable secondary antibodies conjugated with
horseradish peroxidase for 1 h at RT. The immunoreactive bands were
visualized with an enhanced chemiluminescence (ECL) kit (GE
Healthcare, Piscataway, NJ, USA) or the SuperSignal West Femto Kit
(Thermo Scientific). The antibodies used in these studies were as
follows; caspase-3, caspase-8, caspase-9, poly-ADP ribose
polymerase (PARP), Bcl-2, Bax, p53, p21Waf/Cip1, cyclin
B, Cdc2 and β-actin. All primary antibodies were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA) with the exception of
PARP, which was obtained from Enzo Life Sciences (Farmingdale, NY,
USA).
Cell cycle analysis
To compare the effect of EEAR on the HCT-116 cell
cycle, fluorescence-activated cell sorting (FACS) analysis was
performed. Cells were seeded at 1×106 cells/dish in
60-mm culture dishes and incubated for 24 h for attachment. Then,
cells were treated with the indicated concentrations of EEAR for
various time intervals. Detached and attached cells were harvested
and washed with ice-cold PBS. Cells were fixed in ice-cold 70%
(v/v) ethanol and stored at 4°C. For DNA staining, the
ethanol-fixed cells were washed twice with PBS and stained with 50
μg/ml propidium iodide in PBS containing RNase A (500 μg/ml) for 40
min at 37°C. Cell cycle phase distribution was determined with a
FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) by
analyzing at least 10,000 cells per sample.
Real time-polymerase chain reaction
(RT-PCR)
The intracellular p53 mRNA level was determined by
TaqMan RT-PCR. Cells were treated with the indicated concentrations
of drugs for various time intervals, then the total RNAs were
prepared using the easy-spin™ Total RNA Extraction Kit (Intron
Biotechnology, Seoul, Korea). Single-stranded cDNA was synthesized
from 5 μg of total RNA using the SuperScript™ III First-Strand
Synthesis System (Invitrogen). Pre-validated probe and primer sets
for p53 (RefSeq: NM_000546.4, ABI ID Hs01034249_m1, FAM-labeled)
and β-actin endogenous control (RefSeq: NM_001101.3, ABI ID
Hs99999903_m1, VIC-labeled) were purchased from Applied Biosystems
(Foster City, CA, USA). The PCR reactions were run in an Applied
Biosystems Sequence Detection System 7500 and the relative
expression of p53 was calculated using the ΔΔCt method.
Gene silencing of p53
HCT-116 cells were transiently transfected for 24 h
with 400 nM of p53 specific siRNA or green fluorescent protein
(GFP) control siRNA using Lipofectamine 2000 (Invitrogen) according
to the manufacturer’s instructions. The siRNA oligomers were
synthesized by Genolution Pharmaceuticals, Inc. (Seoul, Korea). The
sequences of the tested siRNAs were as follows: 5′-CAG
TCTACCTCCCGCCATA-3′ for p53 and 5′-GGCTACGTC CAGGAGCGCACC-3′ for
the GFP control. The effect of p53 siRNA gene silencing was
determined by western blot analysis.
Statistical analysis
The continuous variables were expressed as mean ±
standard deviation (SD). One-way ANOVA was carried out to compare
the differences between the groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
EEAR-induced caspase-dependent apoptosis
in HCT-116 cells
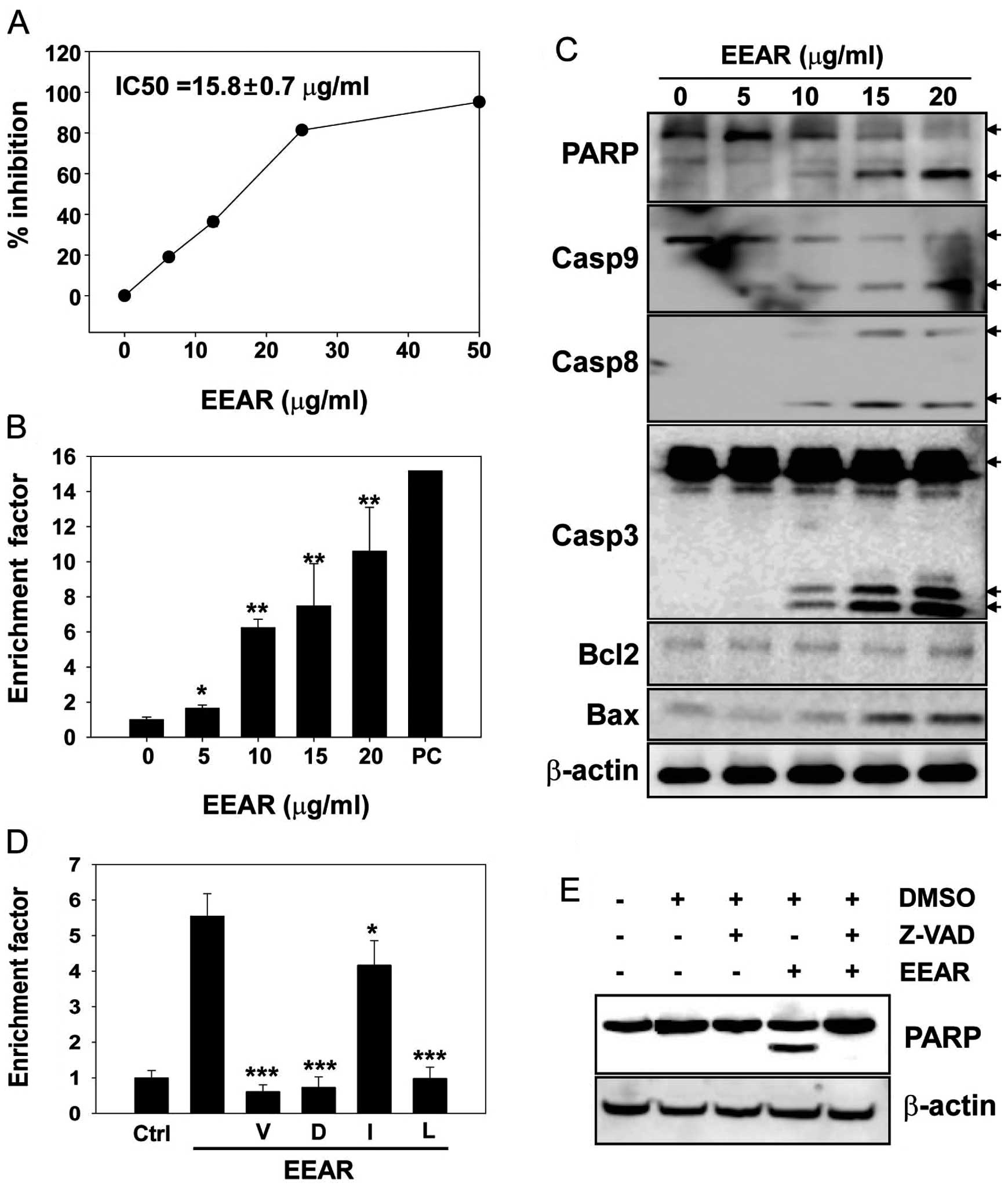
In the present study, the anti-proliferative
potential of EEAR was investigated on HCT-116 cells with an MTS
assay. The HCT-116 cells were exposed to increasing concentrations
of EEAR (0–50 μg/ml) for 48 h and the cell viability was quantified
using an MTS assay kit. The growth of HCT-116 cells was inhibited
by EEAR in a dose-dependent manner and almost 80% growth inhibition
was observed at ≥25 μg/ml EEAR (Fig.
1A). The half-maximal inhibitory concentration (IC50) of EEAR
after 48 h of drug treatment was 15.8±0.68 μg/ml in HCT-116 cells.
According to the US National Cancer Institute (NCI) guideline, a
crude extract and a purified single compound are considered to have
cytotoxic potential if their cytotoxic activities (IC50) are ≤20
and ≤4 μg/ml, respectively, at 48–72 h treatment (11). Based on this guideline, EEAR is
considered cytotoxic in HCT-116 cells in vitro, which
further justified our investigation.
A number of tumor therapeutics with cytotoxic
activity induce apoptosis in cancer cells. To determine whether
EEAR induces apoptosis in HCT-116 cells, apoptosis was quantified
using a commercially available apoptosis detection kit. Cells were
treated with increasing concentrations of EEAR (0–20 μg/ml) for 24
h and apoptosis was quantified using the ELISA method in which
fragmented nucleosomes released from the cells undergoing apoptosis
were detected as described in Materials and methods. Even at the
lowest concentration of EEAR (5 μg/ml), nucleosomes fragmented by
apoptosis were detected and their levels increased in a
dose-dependent manner (Fig. 1B).
Apoptosis caused by treatment with 20 μg/ml EEAR increased 11-fold
compared to the control treatment.
To confirm that EEAR was able to induce apoptosis in
HCT-116 cells, the expression of apoptosis-related proteins was
examined by western blot analysis. HCT-116 cells were treated with
increasing concentrations of EEAR (0–20 μg/ml) for 24 h and each
protein was detected with specific antibodies. The cleavage of the
nuclear protein PARP, a sensitive substrate of active caspase-3,
was observed at ≥10 μg/ml EEAR with parallel activation of caspases
(Fig. 1C). EEAR treatment elevated
the expression of the pro-apoptotic cell death protein, Bax, in a
dose-dependent manner. However, the expression of Bcl2, an
anti-apoptotic cell death protein, was not affected by EEAR
treatment (Fig. 1C).
Since we observed the EEAR-induced activation of
caspases, which play key roles during apoptosis, we determined
whether caspase inhibitors could alleviate EEAR-induced apoptosis
in HCT-116 cells. Pre-treatment with inhibitors of pan-caspases
(z-VAD), caspase-3 (z-DEVD) and caspase-9 (z-LEHD) efficiently
rescued the cells from EEAR-induced apoptosis (Fig. 1D). Although EEAR was observed to
activate caspase-8, which was determined by the cleavage of the
caspase-8 pro-enzyme (Fig. 1C), the
protective effect of the caspase-8 specific inhibitor (z-IETD) was
marginal but statistically significant. The inhibition of caspases
by the pan-caspase inhibitor (z-VAD) was confirmed by complete
inhibition of PARP cleavage induced by EEAR treatment (Fig. 1E). These results revealed that EEAR
induces apoptosis in HCT-116 cells mainly through the intrinsic
apoptotic cell death pathway.
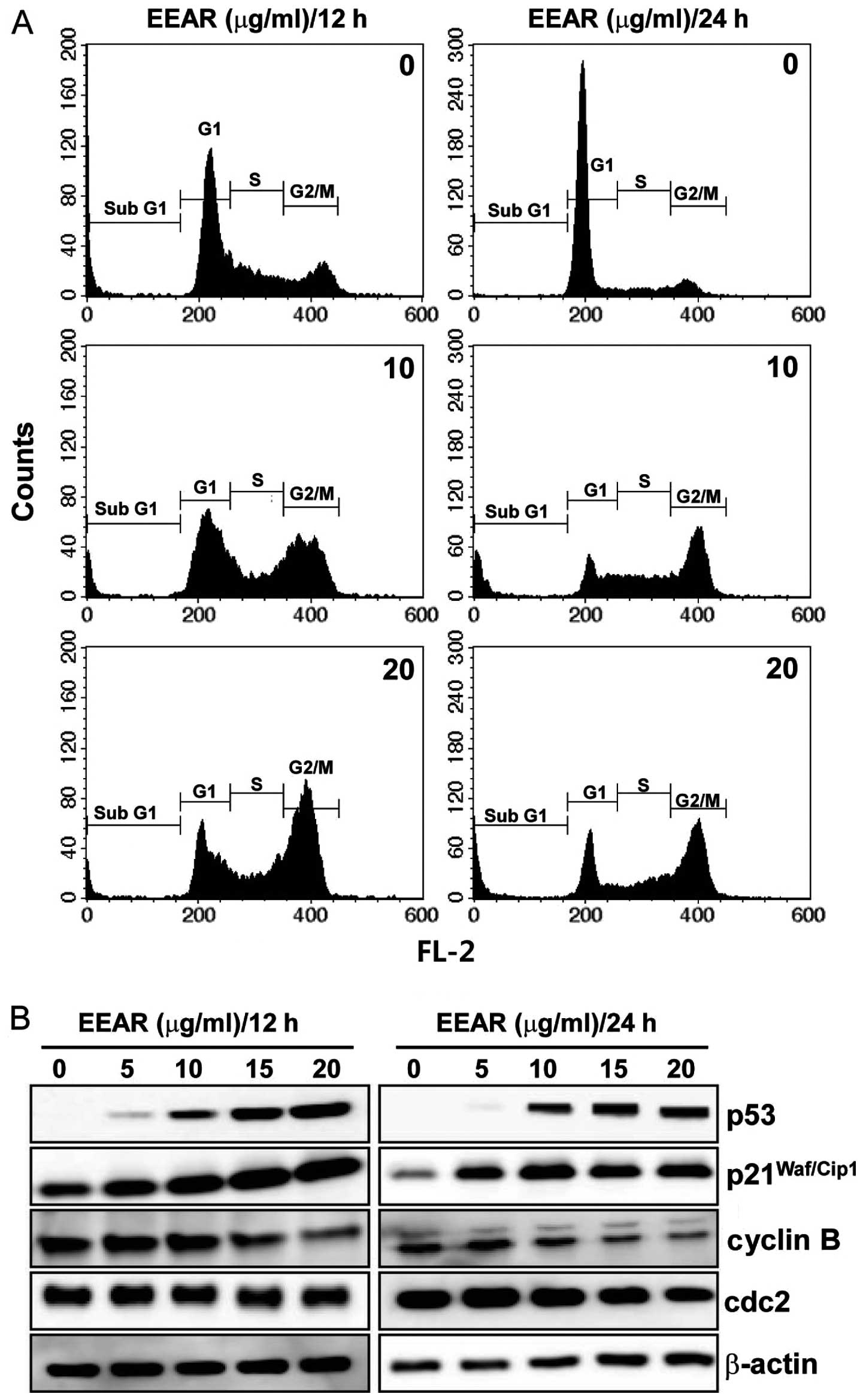
EEAR inhibits cell cycle progression in
HCT-116 cells
A variety of cytotoxic anticancer drugs are known to
affect cell proliferation by disturbing cell cycle progression. To
determine the effect of EEAR on cell cycle progression, HCT-116
cells were treated with EEAR for 12 or 24 h and the intracellular
DNA content was analyzed using a flow cytometer. As shown in
Fig. 2A, EEAR induced cell cycle
arrest in the G2/M phase as early as 12 h. After 24 h, cells
demonstrating subG1 aneuploidy, a hallmark of apoptotic cell death,
accumulated in the presence of EEAR exposure. Since the progression
through the cell cycle at each checkpoint is controlled by specific
regulatory proteins, the changes in the expression of the
regulatory proteins of the G2/M checkpoint were determined by
western blot analysis. Dose-dependent dramatic increases in the p53
and p21Waf/Cip1 tumor suppressors and decreases in the
cyclin B and cdc2 were observed in HCT-116 cells as early as 12 h.
Therefore, EEAR induces G2/M cell cycle arrest in HCT-116 cells by
modulating the expression of the regulatory proteins involved in
the G2/M cell cycle checkpoints.
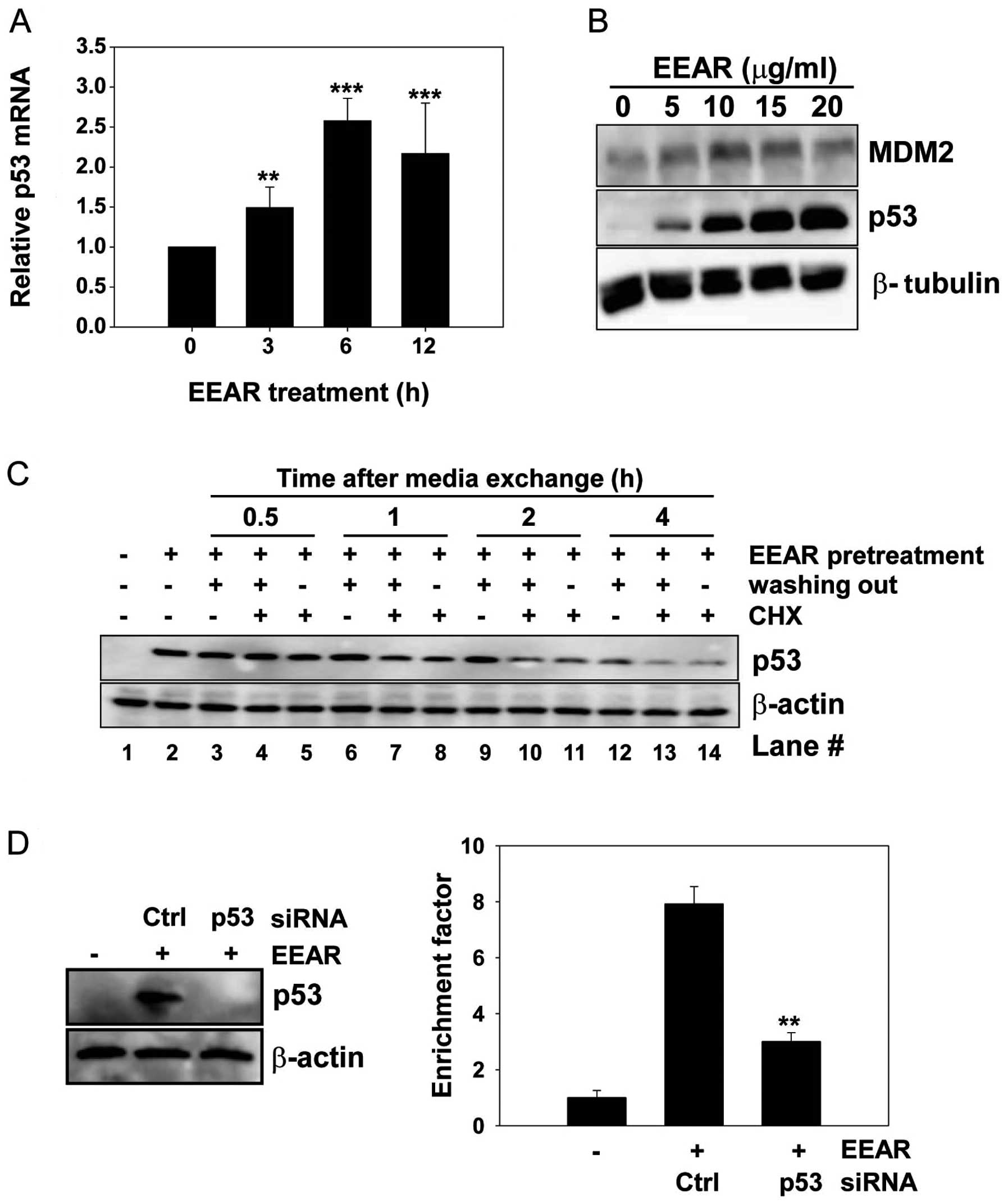
EEAR regulates p53 expression at the
transcription level
Expression of the tumor suppressor protein p53 is
known to be regulated at the transcriptional and post-translational
levels. We carried out quantitative RT-PCR to investigate the
effect of EEAR on the level of intracellular p53 mRNA. Total RNA
was isolated from HCT-116 cells at regular time intervals in the
presence and absence of 10 μg/ml EEAR treatment. The level of p53
mRNA began to rise as early as 3 h after EEAR treatment and reached
a plateau at 6 h (Fig. 3A). MDM2 is
involved in the post-translational regulation of p53 stability.
MDM2 binds to p53 and then recruits it from the nucleus to the
cytosol, where MDM2 facilitates the ubiquitin-mediated protein
degradation of p53. In HCT-116 cells, the MDM2 expression slightly
increased with treatment up to 10 μg/ml EEAR, then it began to
decrease over 10 μg/ml EEAR. This result did not coincide with the
dose-dependent elevation of p53 expression by EEAR (Fig. 2B). Next, we determined whether
elevated expression of p53 by EEAR is due to inhibition of
proteasome-mediated p53 degradation by administration of a protein
synthesis inhibitor, cycloheximide, in the HCT-116 cell culture
media. HCT-116 cells were exposed to 10 μg/ml EEAR and then the
culture media were exchanged with fresh media with (no washing out)
or without (washing out) EEAR, in combination with cycloheximide.
At regular time intervals, total proteins were prepared and
subjected to western blot analysis of p53. Elevated expression of
p53 by EEAR pretreatment was maintained without media exchange
(Fig. 3C, lane 2). In the absence
of both EEAR (washing out) and cycloheximide, p53 was maintained up
to 2 h (lane 3, 6 and 9) and began to decrease at 4 h (lane 12).
Irrespective of the presence of EEAR, cycloheximide significantly
reduced the p53 expression as early as 1 h post-treatment (lane 7
and 8) and almost abolished the p53 expression at 4 h (lane 13 and
14). These results suggest that EEAR does not inhibit
proteasome-mediated p53 degradation. Taken together, the
upregulation of p53 by EEAR was due to the increased level of
intracellular mRNA.
EEAR-induced apoptosis in HCT-116 cells depends on
p53. Since we observed G2/M cell cycle arrest and intracellular
accumulation of p53 upon EEAR treatment, we sought to determine
whether p53 is required for EEAR-induced apoptosis by knocking down
p53 with siRNA. HCT-116 cells were transfected with p53-specific
siRNA or a negative control, GFP siRNA, for 24 h, followed by EEAR
treatment. Transient expression of p53 siRNA efficiently inhibited
the accumulation of p53 induced by EEAR (Fig. 3D, left) and knockdown of p53
conferred resistance to EEAR cytotoxicity in the HCT-116 cells
(Fig. 3D, right). These results
suggest that p53 contributes to EEAR-induced apoptosis.
Discussion
The majority of the medicinal herbs called A.
radixes originate from A. heterotropoides or A.
sieboldii. They have been widely used in traditional Korean
medicine as a component of herb prescriptions due to their diverse
pharmaceutical effects, such as pain killing, anti-allergy and
anti-inflammatory (4,5). However, until recently, few studies
reported the anticancer effect of A. radix(10). To our knowledge, only one mechanism
of the anticancer effect of A. radixes has been suggested by
a Japanese research group. Takara et al revealed that the
whole extract of A. radix increased the substrate transport
function of the multidrug resistance (MDR)/P-glycoprotein, a major
mechanism of MDR and sensitized HeLa cervical cancer cells to
paclitaxel, a well-known MDR/P-glycoprotein substrate, but not to
5-fluorouracil, which is not an MDR/P-glycoprotein substrate
(12). In our study, we evaluated
the in vitro cytotoxic potential of the ethanol extract of
A. radix in HCT-116 human cancer cells and suggested the
potential anticancer mechanism of A. radix.
We demonstrated that EEAR exerted notable
cytotoxicity in a dose-dependent manner and induced apoptosis in
HCT-116 cells (Fig. 1A and B).
Treatment with EEAR increased the expression of Bax and induced
caspase activation (Fig. 1C). The
expression of the anti-apoptotic protein Bcl2 was minimally changed
by EEAR; however, the overall ratio of Bax (pro-apoptosis)/Bcl2
(anti-apoptosis) increased. The increased Bax/Bcl2 ratio is known
to be a rheostat of cell susceptibility to apoptosis (13). Although EEAR activated the caspases
involved in the intrinsic (caspase-9) and extrinsic (caspase-8)
apoptotic pathways (Fig. 1C), more
efficient cell recovery from EEAR cytotoxicity by pretreatment with
the caspase-3 (z-DEVD) and caspase-9 inhibitors (z-LEHD) than the
caspase-8 inhibitor (z-IETD) was observed, suggesting that EEAR may
induce apoptosis in HCT-116 cells mainly through the intrinsic
apoptotic pathway (Fig. 1D).
Intrinsic apoptosis is characterized by an increased mitochondrial
membrane permeability, release of cyt C and mitochondrial
dysfunction. Intracellular reactive oxygen species (ROS) are
produced upon mitochondrial dysfunction and EEAR treatment
increased the intracellular level of ROS in HCT-116 cells (data not
shown). EEAR-induced apoptosis was preceded by a tight cell cycle
arrest in the G2/M phase (Fig. 2A),
which was followed by the downregulation of cdc2 and cyclin B as
well as the upregulation of p53 and p21Waf/Cip1
(Fig. 2B). These results suggest
that EEAR prevents the growth of HCT-116 cells by preventing cell
cycle progress at the G2/M checkpoint, which subsequently leads to
apoptosis.
We demonstrated that EEAR elevates the expression of
p53. As a transcriptional activator, the p53 tumor suppressor has a
broad range of anticancer functions, such as the arrest of cell
growth, induction of apoptosis and senescence and inhibition of
tumorigenic angiogenesis through transcriptional regulation of its
target genes (14–17). Functionally inactive mutations in
the p53 gene or a lack of p53 expression have been observed in a
number of cancers (18–20) and clinical studies have revealed
that over 50% of human tumors carry the p53 mutation (14,21).
In HCT-116 cells, which are known to have wild-type p53, endogenous
p53 was expressed at an undetectable level under normal culture
conditions, but its expression dramatically increased upon EEAR
exposure (Fig. 2B). The
intracellular level of p53 is controlled by post-translational
modification (22) or
transcriptional regulation (23).
The stability of the p53 protein was mainly regulated by the
oncoprotein MDM2, which possesses ubiquitin ligase E3 activity.
MDM2 binds to p53, transports it from the nucleus to the cytosol
and triggers the degradation of p53 via the ubiquitin-proteasome
degradation system. From our study, we suggest that EEAR can
regulate p53 expression at the transcriptional level based on two
observations. First, EEAR increased the p53 mRNA level determined
by RT-PCR (Fig. 3A). Second, the
intracellular protein level of p53 decreased, even in the presence
of EEAR treatment when cells were co-treated with the protein
synthesis inhibitor, cycloheximide (Fig. 3C). However, we cannot exclude the
possibility of an increase in mRNA stability leading to the
elevated p53 levels observed upon EEAR treatment.
Next, we investigated whether p53 is a key modulator
during EEAR-induced apoptosis. The evidence showing that efficient
knockdown of p53 conferred resistance to EEAR cytotoxicity in the
HCT-116 cells (Fig. 3D)
demonstrates that apoptosis induced by EEAR requires the expression
of p53 in HCT-116 cells. The apoptosis mediated by p53 can be
either dependent on, or independent of its transcriptional
activity. In the transcription-dependent pathway, transcriptionally
active p53 upregulates the expression of pro-apoptotic proteins,
such as Bax and p53 upregulated modulator of apoptosis (PUMA),
thereby inhibiting the anti-apoptotic survival protein Bcl2. In the
transcription-independent pathway, p53 cooperates with other
apoptotic factors, such as E2F-1 and induces apoptosis irrespective
of its transcriptional activity (24). In the present study, we could not
determine which pathway the HCT-116 cells follow after EEAR
treatment.
In this study, we demonstrated that the ethanol
extract of A. radix induces G2/M cell cycle arrest and
apoptosis in HCT-116 human colon cancer cells in vitro
through upregulation of the p53 tumor suppressor. This study
increases our understanding of the cytotoxic mechanisms of EEAR and
suggests that EEAR contains natural herb materials that could be
used in the development of anticancer drugs. The active compounds
responsible for the cytotoxic effect of the ethanol extract of
A. radix will be identified in our future studies.
Acknowledgements
The authors sincerely thank Dr Go Ya
Choi, Dr Sungwook Chae and Hye Won Lee, Herbal Medicine Research
Division, for identification of the herbal plants and preparation
of herbal extract. This research was supported by a grant from the
Korea Institute of Oriental Medicine (KIOM, K12061).
References
|
1
|
Wang CZ, Luo X, Zhang B, et al:
Notoginseng enhances anti-cancer effect of 5-fluorouracil on human
colorectal cancer cells. Cancer Chemother Pharmacol. 60:69–79.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen J, Huang XF and Katsifis A:
Activation of signal pathways and the resistance to anti-EGFR
treatment in colorectal cancer. J Cell Biochem. 111:1082–1086.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kinzler KW and Vogelstein B: Lessons from
hereditary colorectal cancer. Cell. 87:159–170. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jang JY, Lee JH, Shin HK, Choi YH, Lee JD
and Choi BT: Partially purified Asiasari radix inhibits
melanogenesis through extracellular signal-regulated kinase
signaling in B16F10 cells. Int J Mol Med. 25:287–292. 2010.
|
|
5
|
Suzuki Y, Yuzurihara M, Hibino T, Yano S
and Kase Y: Aqueous extract of Asiasari radix inhibits
formalin-induced hyperalgesia via NMDA receptors. J Ethnopharmacol.
123:128–133. 2009.
|
|
6
|
Kim HM and Moon YS: Asiasari radix
inhibits immunoglobulinE production on experimental models in
vitro and in vivo. Immunopharmacol Immunotoxicol.
21:469–481. 1999. View Article : Google Scholar
|
|
7
|
Hashimoto K, Yanagisawa T, Okui Y, Ikeya
Y, Maruno M and Fujita T: Studies on anti-allergic components in
the roots of Asiasarum sieboldi. Planta Med. 60:124–127.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han Y, Kwon EH and Kim SJ: Protection of
brain cells against AMPA-induced damage by Asiasari Radix
extracts. Phytother Res. 17:882–886. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han Y and Kim SJ: Memory enhancing actions
of Asiasari radix extracts via activation of insulin
receptor and extracellular signal regulated kinase (ERK) I/II in
rat hippocampus. Brain Res. 974:193–201. 2003.PubMed/NCBI
|
|
10
|
Takasaki M, Konoshima T, Yasuda I, Hamano
T and Tokuda H: Inhibitory effects of shouseiryu-to on two-stage
carcinogenesis. II. Anti-tumor-promoting activities of lignans from
Asiasarum heterotropoides var mandshuricum. Biol Pharm Bull.
20:776–780. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ramasamy S, Abdul Wahab N, Zainal Abidin
N, Manickam S and Zakaria Z: Growth inhibition of human gynecologic
and colon cancer cells by Phyllanthus watsonii through
apoptosis induction. PLoS One. 7:e347932012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takara K, Horibe S, Obata Y, Yoshikawa E,
Ohnishi N and Yokoyama T: Effects of 19 herbal extracts on the
sensitivity to paclitaxel or 5-fluorouracil in HeLa cells. Biol
Pharm Bull. 28:138–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Korsmeyer SJ, Shutter JR, Veis DJ, Merry
DE and Oltvai ZN: Bcl-2/Bax: a rheostat that regulates an
anti-oxidant pathway and cell death. Semin Cancer Biol. 4:327–332.
1993.PubMed/NCBI
|
|
14
|
Pei D, Zhang Y and Zheng J: Regulation of
p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 3:228–235.
2012.PubMed/NCBI
|
|
15
|
Futamura M, Kamino H, Miyamoto Y, et al:
Possible role of semaphorin 3F, a candidate tumor suppressor gene
at 3p21.3, in p53-regulated tumor angiogenesis suppression. Cancer
Res. 67:1451–1460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tanikawa C, Nakagawa H, Furukawa Y,
Nakamura Y and Matsuda K: CLCA2 as a p53-inducible senescence
mediator. Neoplasia. 14:141–149. 2012.PubMed/NCBI
|
|
17
|
Rahman-Roblick R, Roblick UJ, Hellman U,
et al: p53 targets identified by protein expression profiling. Proc
Natl Acad Sci U S A. 104:5401–5406. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Braithwaite AW, Royds JA and Jackson P:
The p53 story: layers of complexity. Carcinogenesis. 26:1161–1169.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
El-Deiry WS: The role of p53 in
chemosensitivity and radiosensitivity. Oncogene. 22:7486–7495.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reisman D and Loging WT: Transcriptional
regulation of the p53 tumor suppressor gene. Semin Cancer Biol.
8:317–324. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Honda R, Tanaka H and Yasuda H:
Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53.
FEBS Lett. 420:25–27. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun X, Shimizu H and Yamamoto K:
Identification of a novel p53 promoter element involved in
genotoxic stress-inducible p53 gene expression. Mol Cell Biol.
15:4489–4496. 1995.PubMed/NCBI
|
|
24
|
Haupt S, Berger M, Goldberg Z and Haupt Y:
Apoptosis - the p53 network. J Cell Sci. 116:4077–4085. 2003.
View Article : Google Scholar : PubMed/NCBI
|