Introduction
Glutathione S-transferases (GSTs), a superfamily of
detoxifying enzymes, contain at least five subclasses, including α,
μ, π, ω and θ. GSTs act catalytically through the
nucleophilic attachment of the sulfur atom of glutathione (GSH)
onto the electrophilic groups of substrate molecules (1,2). GSTs
are important in protecting cells from cytotoxic and carcinogenic
agents, removing oxidative stress products, and modulating cell
proliferation and signaling pathways (2,3). As an
isozyme of GST, GSTP1 is a major regulator of cell signaling in
response to stress, hypoxia, growth factors and other stimuli.
Previous studies have demonstrated that GSTP1 inhibits
lipopolysaccharide-induced MAPK, and that NF-κB activation
decreases LPS-induced iNOS production by regulating MAPK activation
(4). In addition, GSTP1 expression
is highly correlated with carcinogenesis; GSTP1 is overexpressed in
a variety of human cancers, including lung, colon, ovary, bladder
and kidney cancer (5–8). By contrast, the reduced expression and
activity of GSTP1 are observed due to the hypermethylation of its
promoter in hepatocellular carcinoma (HCC) and prostate cancer
(9–10), although GSTP1 may also be detected
in the corresponding non-tumorous tissues. However, GSTP1 null mice
reveal an increased risk of carcinogen-induced skin tumorigenesis
(11). Notably, the overexpression
of GSTP1 has been reported to protect prostate cells from
cytotoxicity and DNA damage due the heterocyclic amine carcinogen
PhIP (12), which suggests that
silencing of the GSTP1 gene by CpG island DNA methylation may be
important in the development of HCC.
The signal transducer and activator of transcription
(Stat) family of cytoplasmic proteins is important for promoting
the proliferation, survival, and other biological processes
triggered by cytokines and growth factors, including epidermal
growth factor (EGF) (13–15). EGF induces the activation of Stat1,
Stat3 and Stat5 in cancer cells. Stat3 has been demonstrated to
play a critical role in EGF signaling in both normal and tumor
cells (16). Normal Stat activation
is a highly regulated process. However, atypical activation of
Stat3 is usually detected in various human tumors including HCC,
and may modulate the oncogenic transformation and progression
(17). Furthermore, Stat3 has been
implicated as a promising target for HCC therapy, as the inhibition
of Stat3 has been shown to induce growth arrest and apoptosis of
human HCC cells (18). Since GSTP1
exerts important anti-inflammatory, antioxidant and detoxification
functions in the body, and its promoter is hypermethylated in HCC,
the restoration of GSTP1 expression may be a promising method for
preventing tumors. In the present study, the possible regulatory
mechanisms of GSTP1 on Stat activation have been explored in HepG2
cells. The results indicate that the overexpression of GSTP1
specifically downregulates Stat3 activation, and inhibits cell
growth via a direct interaction between GSTP1 and Stat3.
Materials and methods
Antibodies and reagents
The p-Stat3 (Y705), Stat3 and cyclin D1 antibodies
were purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA). A mouse monoclonal antibody against xpress-tag was purchased
from Invitrogen Life Technologies (Carlsbad, CA, USA). Flag-tag was
purchased from Sigma (St. Louis, MO, USA). GAPDH and protein G were
purchased from Roche Applied Science (Indianapolis, IN, USA). Mouse
or rabbit A/G and IgG antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Secondary antibodies
coupled to IRDye 800 fluorophore for the Odyssey Infrared Imaging
System were purchased from Rockland Immunochemicals, Inc.
(Gilbertsville, PA, USA).
Plasmid construction
Flag-Stat3 (wt) was provided by Dr Zhijie Chang of
Tsinghua University (Beijing, China). GSTP1-RNAi was constructed
into pRNA-u6. All expression plasmids were confirmed by sequencing
and purified by the Endofree Plasmid Preparation kit (Qiagen,
Hilden, Germany).
Cell culture and transfection
HEK293, HepG2 and WRL-68 cell lines were purchased
from the Institute of Biochemistry and Cell Biology, the Chinese
Academy of Sciences (Shanghai, China), and then cultured in
Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen Life
Technologies) supplemented with 10% fetal bovine serum (FBS;
HyClone Laboratories; Logan, UT, USA), 100 U/ml penicillin and 100
μg/ml streptomycin in 5% CO2 at 37°C. Transient
transfection was performed using the Lipofectamine 2000 reagent
(Invitrogen Life Technologies) according to the manufacturer’s
instructions. In all cases, the total amount of DNA was normalized
by the empty control plasmids.
Immunoprecipitation and immunoblotting
analysis
HEK293 cells were washed twice with ice-cold
phosphate-buffered saline (PBS; pH 7.4) and lysed in lysis buffer
containing 20 mM Tris (pH 7.5), 135 mM NaCl, 2 mM EDTA, 2 mM
dithiothreitol (DTT), 25 mM β-glycerophosphate, 2 mM sodium
pyrophosphate, 10% glycerol, 1% Triton X-100, 1 mM sodium
orthovanadate, 10 mM NaF and 1 mM phenylmethylsulfonyl fluoride
(PMSF), supplemented with complete protease inhibitor cocktail
(Roche Applied Science). Following incubation on ice for 30 min,
the cell lysates were centrifuged at 15,000 × g at 4°C for 15 min.
Proteins (500 μg) were immunoprecipitated with the
designated antibodies, respectively. The precleared Protein A/G
PLUS-Agarose beads (Santa Cruz Biotechnology, Inc.) were incubated
with immunocomplexes for 2 h and washed four times with the lysis
buffer. The immunoprecipitates were subjected to sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), then
transferred onto a nitrocellulose membrane (Hybond-C; Amersham
Biosciences Corp.; Piscataway, NJ, USA). The immunoblotting
analyses were performed. The results were visualized using IRDye
800 fluorophore-conjugated antibody in the Li-COR Odyssey Infrared
Imaging System according to the manufacturer’s instructions (LI-COR
Biosciences; Lincoln, NE, USA).
Cell cycle assay
Cells were collected by trypsinization, pelleted at
800 × g for 10 min and fixed in 70% ethanol. The DNA content was
evaluated by flow cytometry with propidium iodide (PI) staining.
Flow cytometric analysis was performed using FACScan
(Becton-Dickinson; Mountain View, CA, USA) with Cell Quest
software.
Cell viability
The transfected HepG2 cells were seeded in 96-well
plates and the cell viability was evaluated by a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. For each experiment, six wells were used and the experiments
were repeated three times.
Statistical analysis
All experimental data was obtained from cultured
cells were expressed as mean ± SD. Western blotting analysis
experiments were repeated 3 times with similar trends. A one-way
repeated measure analysis ofvariance and a Student’s t-test were
used to determine the significance of the difference between two
groups.
Results
Overexpression of GSTP1 inhibits
EGF-induced Stat3 activation
In order to explore the effect of GSTP1 on
endogenous Stat3 activation in HepG2 cells, the cells were
transfected with xpress-tagged GSTP1 followed by EGF stimulation.
The phosphorylation of Stat3 was examined by western blot analysis.
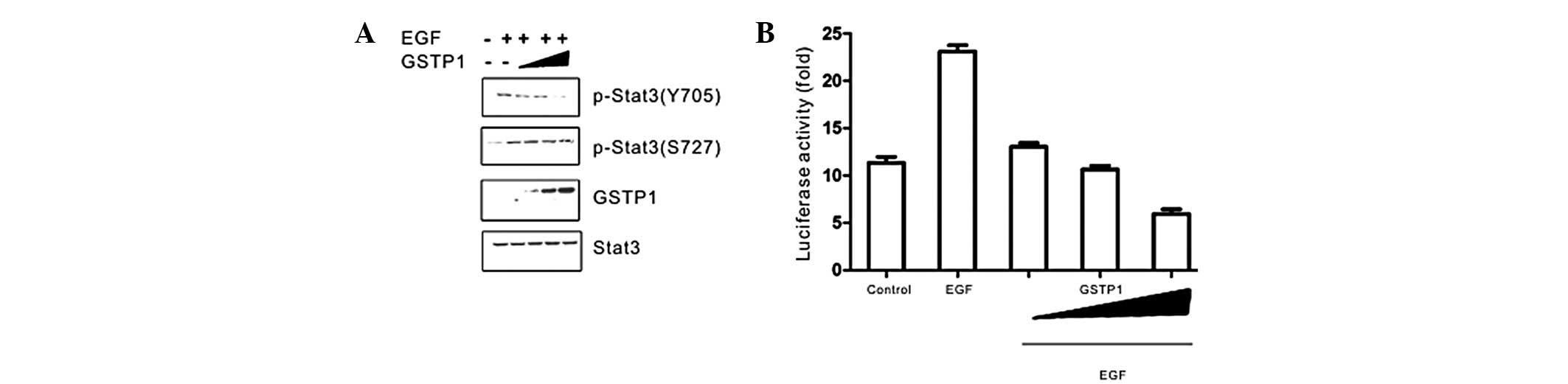
Overexpression of GSTP1 inhibited the EGF-stimulated tyrosine
phosphorylation of Stat3 in a dose-dependent manner (Fig. 1A). However, serine phosphorylation
of Stat3 and a change in the expression of Stat3 were not observed.
In order to explore whether GSTP1 is able to modulate Stat3
transcriptional activity in the presence of EGF, HepG2 cells were
co-transfected with Stat3-dependent luciferase reporter gene and an
xpress-GSTP1 plasmid. As is demonstrated in Fig. 1B, the cells were stimulated by EGF
for 15 min and a 3-fold enhancement in fluorescence intensity was
observed when compared with the control cells. However, the
increase of fluorescence intensity was blocked in the presence of
exogenous GSTP1. These results indicate that the suppression of
Stat3 transcription may result from the inhibition of its tyro-sine
phosphorylation.
GSTP1 knockdown increases tyrosine
phosphorylation of Stat3 stimulated by EGF
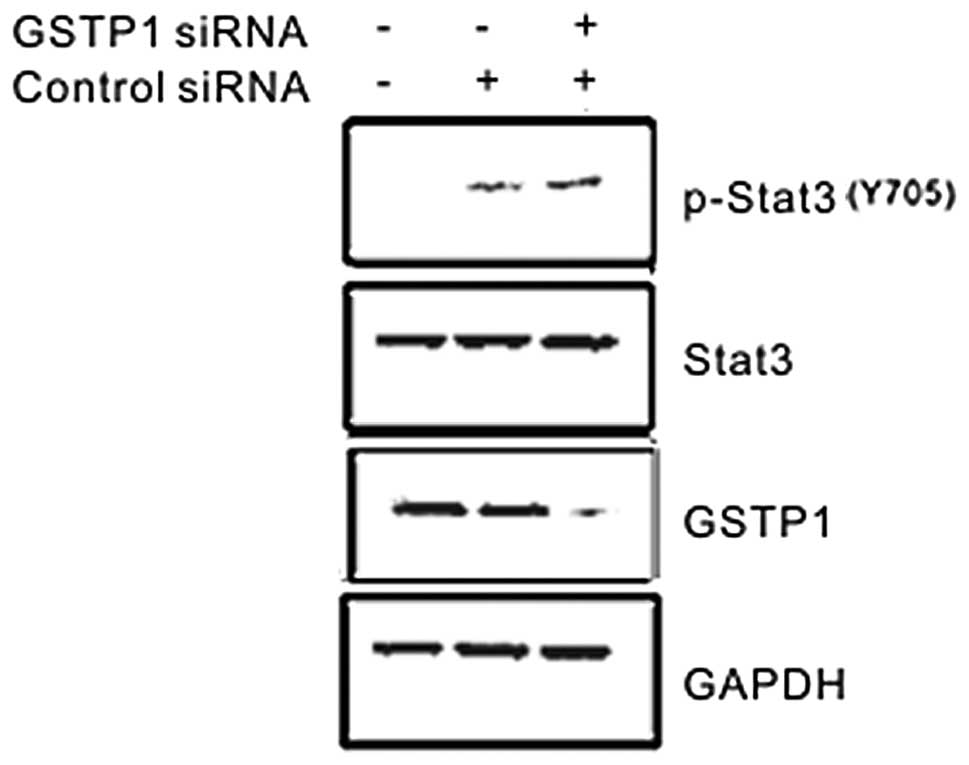
In order to further confirm whether GSTP1
downregulated the phosphorylation of Stat3, the GSTP1 siRNA was
transfected into WRL-68 cells that have higher endogenous levels of
Stat3. The effect of GSTP1 siRNA on EGF-mediated tyrosine
phosphorylation of Stat3 was examined. As expected, the expression
of GSTP1 was effectively blocked by GSTP1 siRNA, and GSTP1 siRNA
further enhanced the EGF-stimulated tyrosine phosphorylation of
Stat3 (Fig. 2). By contrast, GSTP1
siRNA had no effect on the expression level of Stat3. These results
indicated that endogenous GSTP1 negatively regulated EGF-induced
Stat3 activation.
Effects of forced expression of GSTP1 on
the cell proliferation and cell cycle phase distribution in HepG2
cells
Since GSTP1 inhibits Stat3-dependent luciferase
activity and previous studies have demonstrated that GSTP1 inhibits
cell proliferation (19), the
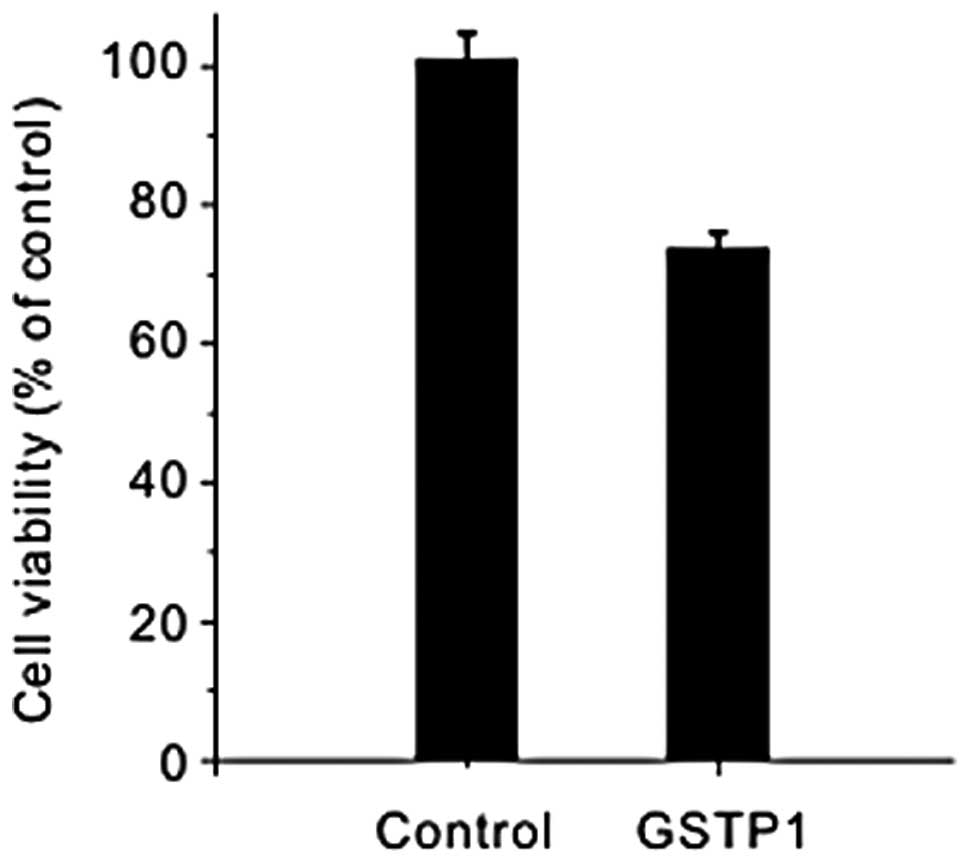
effect of GSTP1 on cell viability was also examined. The HepG2
cells were transfected with 2 μg GSTP1 for 36 h and the
GSTP1-transfected cells exhibited a reduced proliferation compared
with the control HepG2 cells (Fig.
3), which suggested that GSTP1 possessed the function of
suppressing cell proliferation. Additionally, Stat3 is important in
cell cycle progression, whereas the inhibition of
constitutively-active Stat3 induces cell cycle arrest (20,21).
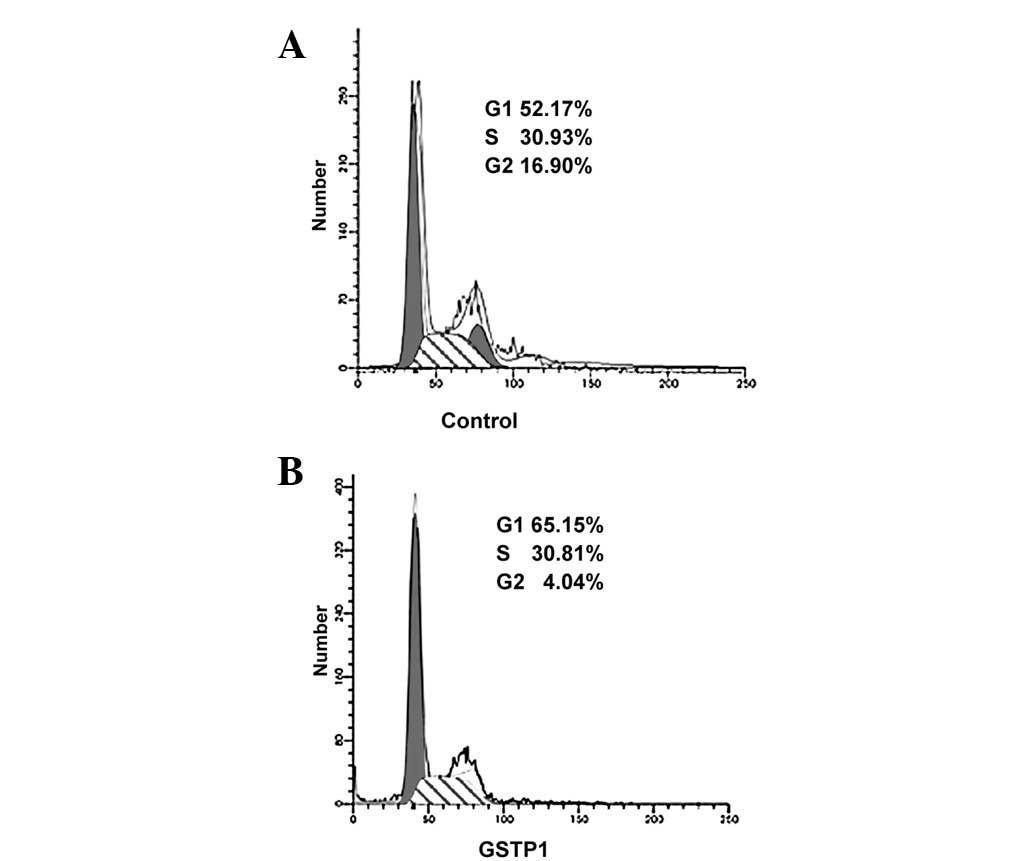
Flow cytometric analyses (Fig. 4)
revealed that overexpression of GSTP1 induces cell cycle arrest and
cell accumulation at the G0–G1 phase by 36 h when compared with
untransfected cells. These results suggest that the GSTP1-induced
inhibition of Stat3 signaling may result in the inhibition of cell
growth and blockage of the cell cycle.
GSTP1 expression inhibits Stat3 but not
its upstream regulators
It is well known that the phosphorylation of Stats
depends on the activation of JAKs and/or Src family kinases, which
stimulated us to explore the possible tyrosine kinases involved in
the activation of Stats through GSTP1 inhibition. In order to gain
insights into the inhibitory mechanism of GSTP1 on the Stat
signaling cascade, the effect of GSTP1 on JAK and Src kinase
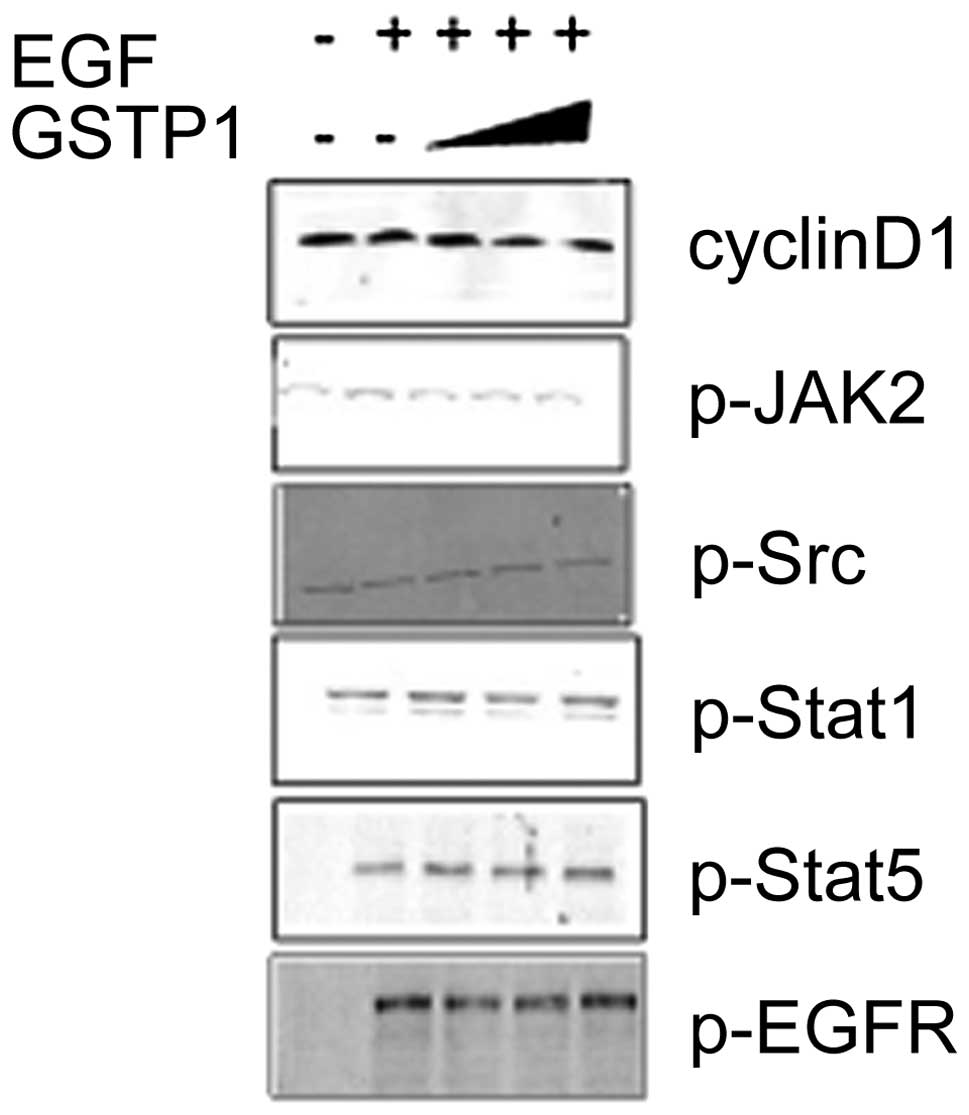
activity was also examined. As demonstrated in Fig. 5, GSTP1 suppressed Stat3-mediated
downstream factor cyclin D1, and did not affect the upstream
regulators, such as p-JAK2, p-Src and p-EGFR, of the
phosphorylation of Stat3. In addition, the phosphorylation of Stat5
was not affected by GSTP1, which revealed the specificity of GSTP1
for the phosphorylation of Stat3. Therefore, GSTP1 may inhibit the
phosphorylation of Stat3 by direct inhibition at its protein
level.
Inhibition of Stat3 activity involves a
direct interaction between GSTP1 and Stat3
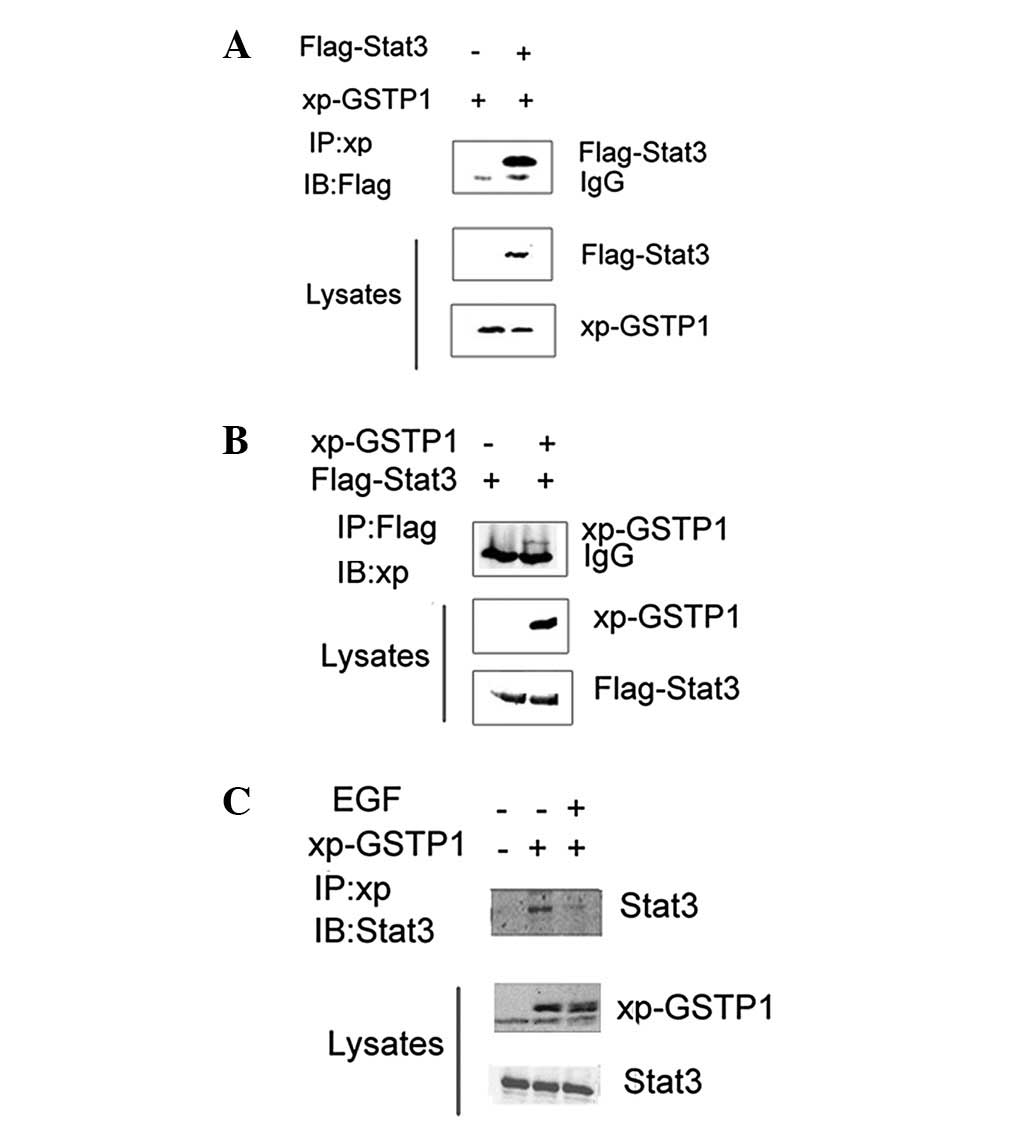
In order to understand the mechanism of suppression
of Stat3 activation, HEK293 cells transiently co-transfected with
Flag-Stat3 and Xpress-GSTP1 were subjected to immunoprecipitation
with anti-Flag or -Xpress antibody to explore the association
between GSTP1 and Stat3. The immunoprecipitates were separated by
SDS-PAGE, and transferred to a nitrocellulose membrane. The results
indicated that GSTP1 co-immunoprecipitated with Stat3 in HEK293
cells (Figs. 6A and B). Similarly,
a specific association between GSTP1 and endogenous Stat3 was
observed in Xpress-GSTP1-transfected HepG2 cells. The
co-immunoprecipitation assay demonstrated that Xpress-GSTP1 was
able to physically interact with endogenous Stat3 (Fig. 6C). These results suggest that the
negative regulatory effect of GSTP1 on Stat3 is mediated by a
physical interaction between the two proteins.
Discussion
GSTs are a superfamily of detoxifying enzymes that
catalyze the conjugation of reduced GSH via a variety of
electrophiles. In addition to their catalytic functions, GSTs also
serve as nonenzymatic binding proteins, interacting with various
lipophilic compounds including steroid and thyroid hormones
(22–25). Furthermore, GSTP1 also regulates
important normal cellular functions through its interaction with a
number of critical cellular proteins, such as transglutaminase 2
(TGM2), apoptosis signal-regulating kinase 1 (ASK1) and Fanconi
anemia group C protein (FANCC) (25). These findings suggest that the
diverse functions of GSTP1 may be determined by the interactions
with its key partner proteins. The currently identified mechanisms
of Stat3 inhibition include dephosphorylation, inactivation of JAK
by suppressor of cytokine signaling (SOCS1) protein (26) and abrogation of DNA binding by the
protein inhibitor of activated Stat (PIAS) (27). However, in the present study, the
physical interaction between GSTP1 and Stat3 resulted in the
suppression of Stat3 activity (Fig.
6). Notably, upon treatment of HepG2 cells with EGF, a
disassociation of the GSTP1/Stat3 complex was achieved, suggesting
that Stat3 forms a complex with GSTP1 and that EGF may release
Stat3 from the GSTP1 binding complex. The mechanism whereby GSTP1
specifically interacts with Stat3 may present a novel means of
therapeutic intervention in Stat3-driven tumors.
HCC is the most common type of primary liver cancer,
particularly in developing countries. More than half of cancer
patients are identified as having HCC in China (28). Previous studies have reported that
Stat3 is a promising target for HCC therapy (18). GSTP1 is downregulated or absent in
HCC due to the action of DNA methyltransferase. However, the
clinical applications of nucleoside analogs used as DNA
methyltransferase inhibitors are limited somewhat by
myelosuppression and other potential side effects. In the present
study, the restoration of the GSTP1 protein has been demonstrated
to exert an anticancer effect by inhibiting the Stat3 signaling
pathway in HCC cells. We have demonstrated that the overexpression
of GSTP1 in HepG2 cells suppresses the tyrosine phosphorylation and
transcription activity of EGF-inducible Stat3, as well as the gene
expression of Stat3-regulated cyclin D1, thus resulting in the
inhibition of proliferation and increased accumulation of cells in
the G1/G0 phase (Fig. 4). JAK2 or
Src may be common upstream effectors for the activation of Stat3.
Our results demonstrated that GSTP1 only inhibited Stat3, but no
JAK2-dependent mechanism was observed in the transfected cells
(Fig. 5).
In summary, a novel function of GSTP1 in inhibiting
EGF-induced Stat3 activation has been demonstrated. The GSTP1-Stat3
complex reduces proliferation and arrests the cell cycle by
terminating Stat3 activity.
Acknowledgements
The authors would like to thank Dr
Zhijie Chang (Tsinghua University) for providing the plasmid
constructs used in this study. This study was supported by grants
from the National Nature Science Foundation of China (Nos. 30770842
and 30771979).
References
|
1
|
Tew KD: Glutathione-associated enzymes in
anticancer drug resistance. Cancer Res. 54:4313–4320.
1994.PubMed/NCBI
|
|
2
|
Townsend D and Tew K: Cancer drugs,
genetic variation and the glutathione-S-transferase gene family. Am
J Pharmacogenomics. 3:157–172. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eaton DL and Bammler TK: Concise review of
the glutathione S-transferases and their significance to
toxicology. Toxicol Sci. 49:156–164. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xue B, Wu Y, Yin Z, Zhang H, Sun S, Yi T
and Luo L: Regulation of lipopolysaccharide-induced inflammatory
response by glutathione S-transferase P1 in RAW264.7 cells. FEBS
Lett. 579:4081–4087. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Inoue T, Ishida T, Suqio K, Maehara Y and
Suqimachi K: Glutathione S transferase Pi is a powerful indicator
in chemotherapy of human lung squamous-cell carcinoma. Respiration.
62:223–227. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sato K: Glutathione transferases as
markers of preneoplasia and neoplasia. Adv Cancer Res. 52:205–255.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singh SV, Xu BH, Gupta V, Emerson EO,
Zaren H and Jani JP: Characterization of a human bladder cancer
cell line selected for resistance to BMY 25067, a novel analogue of
mitomycin C. Cancer Lett. 95:49–56. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grignon DJ, Abdel-Malak M, Mertens WC,
Sakr WA and Shepherd RR: Glutathione S-transferase expression in
renal cell carcinoma: a new marker of differentiation. Mod Pathol.
7:186–189. 1994.PubMed/NCBI
|
|
9
|
Zhong S, Tang MW, Yeo W, Liu C, Lo YM and
Johnson PJ: Silencing of GSTP1 gene by CpG island DNA
hypermethylation in HBV-associated hepatocellular carcinomas. Clin
Cancer Res. 8:1087–1092. 2002.PubMed/NCBI
|
|
10
|
Lin X, Asgari K, Putzi MJ, et al: Reversal
of GSTP1 CpG island hypermethylation and reactivation of pi-class
glutathione S-transferase (GSTP1) expression in human prostate
cancer cells by treatment with procainamide. Cancer Res.
61:8611–8616. 2001.PubMed/NCBI
|
|
11
|
Henderson CJ, Smith AG, Ure J, Brown K,
Bacon EJ and Wolf CR: Increased skin tumorigenesis in mice lacking
pi class glutathione S-transferases. Proc Natl Acad Sci USA.
95:5275–5280. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nelson CP, Kidd LC, Sauvageot J, et al:
Protection against
2-hydroxyamino-1-methyl-6-phenylimidazo[4,5-b]pyridine cytotoxicity
and DNA adduct formation in human prostate by glutathione
S-transferase P1. Cancer Res. 61:103–109. 2001.
|
|
13
|
Bromberg J and Darnell JE Jr: The role of
STATs in transcriptional control and their impact on cellular
function. Oncogene. 19:2468–2473. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Darnell JE Jr: Transcription factors as
targets for cancer therapy. Nat Rev Cancer. 2:740–749. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang T, Ma J and Cao X: Grb2 regulates
Stat3 activation negatively in epidermal growth factor signalling.
Biochem J. 376:457–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Buettner R, Mora LB and Jove R: Activated
STAT signaling in human tumors provides novel molecular targets for
therapeutic intervention. Clin Cancer Res. 8:945–954.
2002.PubMed/NCBI
|
|
18
|
Li F, Fernandez PP, Rajendran P, Hui KM
and Sethi G: Diosgenin, a steroidal saponin, inhibits STAT3
signaling pathway leading to suppression of proliferation and
chemosensitization of human hepatocellular carcinoma cells. Cancer
Lett. 292:197–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ruscoe JE, Rosario LA, Wang T, et al:
Pharmacologic or genetic manipulation of glutathione S-transferase
P1-1 (GSTpi) influences cell proliferation pathways. J Pharmacol
Exp Ther. 298:339–345. 2001.PubMed/NCBI
|
|
20
|
Garcia R, Bowman TL, Niu G, et al:
Constitutive activation of Stat3 by the Src and JAK tyrosine
kinases participates in growth regulation of human breast carcinoma
cells. Oncogene. 20:2499–2513. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Niu G, Bowman T, Huanq M, et al: Roles of
activated Src and Stat3 signaling in melanoma tumor cell growth.
Oncogene. 21:7001–7010. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ishigaki S, Abramovitz M and Listowsky I:
Glutathione-S-transferases are major cytosolic thyroid hormone
binding proteins. Arch Biochem Biophys. 273:265–272. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ketley JN, Habig WH and Jakoby WB: Binding
of nonsubstrate ligands to the glutathione S-transferases. J Biol
Chem. 250:8670–8673. 1975.PubMed/NCBI
|
|
24
|
Litwack G, Ketterer B and Arias IM:
Ligandin: a hepatic protein which binds steroids, bilirubin,
carcinogens and a number of exogenous organic anions. Nature.
234:466–467. 1971. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lo HW and Ali-Osman F: Genetic
polymorphism and function of glutathione S-transferases in tumor
drug resistance. Curr Opin Pharmacol. 7:367–374. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Naka T, Narazaki M, Hirata M, et al:
Structure and function of a new STAT-induced STAT inhibitor.
Nature. 387:924–929. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chung CD, Liao J, Liu B, Rao X, Jav P,
Berta P and Shuai K: Specific inhibition of Stat3 signal
transduction by PIAS3. Science. 278:1803–1805. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|