Introduction
Intracranial manifestations correlated with
autosomal dominant polycystic kidney disease (ADPKD), including
intraparenchymal hemorrhage and aneurysm formation, have been
widely described (1), as has an
increased incidence of intracranial arachnoid cysts (1,2).
Hepatic cysts are relatively common in patients with ADPKD
(3,4), increasing with advancing age, female
gender, pregnancy and degree of renal lesions, which affect the
course of polycystic liver disease (PCLD) (5). The majority of patients with multiple
liver cysts are asymptomatic, recognized only after routine
ultrasound investigation. However, the most common clinical
patterns include abdominal pain, dyspnea and early satiety related
to a compartmental syndrome. In the present study, we described a
rare association of PCLD with intracranial meningiomas in patients
included on a liver transplant waiting list, focusing on the
diagnosis, possible association with any chromosomal alteration and
mandatory treatment of those lesions prior to transplantation.
Case reports
Two female patients, 39 and 49 years of age, were
referred for evaluation for extensive abdominal tumors, with
symptoms related to a compartmental syndrome, which included
dyspnea, abdominal pain and early satiety. The two patients
provided informed consent, and the study was approved by the ethics
committee of Sao Paulo State University.
Clinical investigation included an abdominal
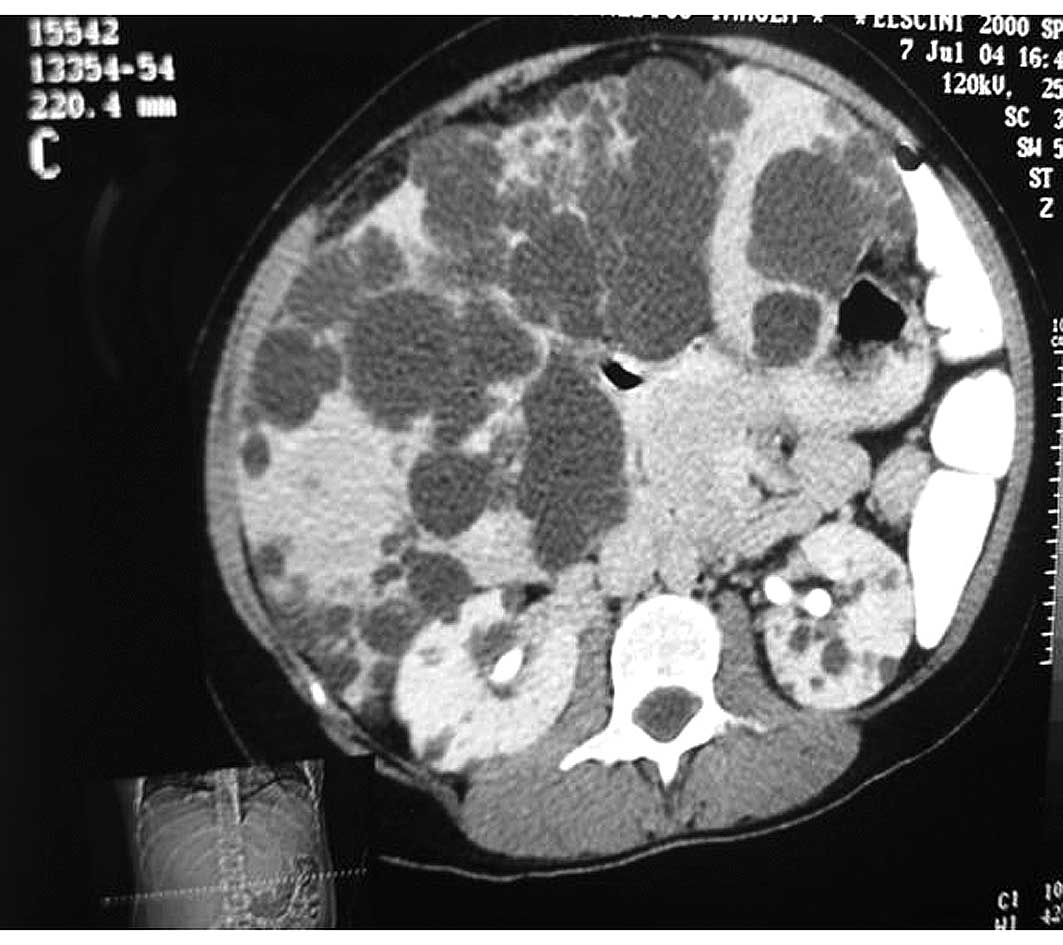
ultrasound and further computer tomography (CT), which revealed an
extensive liver involvement by cyst lesions, without kidney
involvement (Fig. 1). The mean cyst
diameters of the largest liver cysts were 10±5 cm, located deeply
in the liver parenchyma and the small ones, superficially.
Abnormal physical findings were similar in the two
patients, and included right upper quadrant pain and abdominal
distension.
Laboratorial data were collected and included
creatinine, urea nitrogen, aspartate aminotransferase (AST),
alanine aminotransferase (ALT), alkaline phosphatase (ALP),
γ-glutamyl transferase (GGT), total bilirubin, RNI and prothrombin
time, which were relatively stable and without significant
abnormalities for the two patients. The same blood type, AB, was
identified.
The main reasons for including the two patients on a
liver transplant waiting list were the great liver size, which
reached the lower abdominal quadrant, with diaphragmatic and
inferior vena cava compression, leading to edema of the legs.
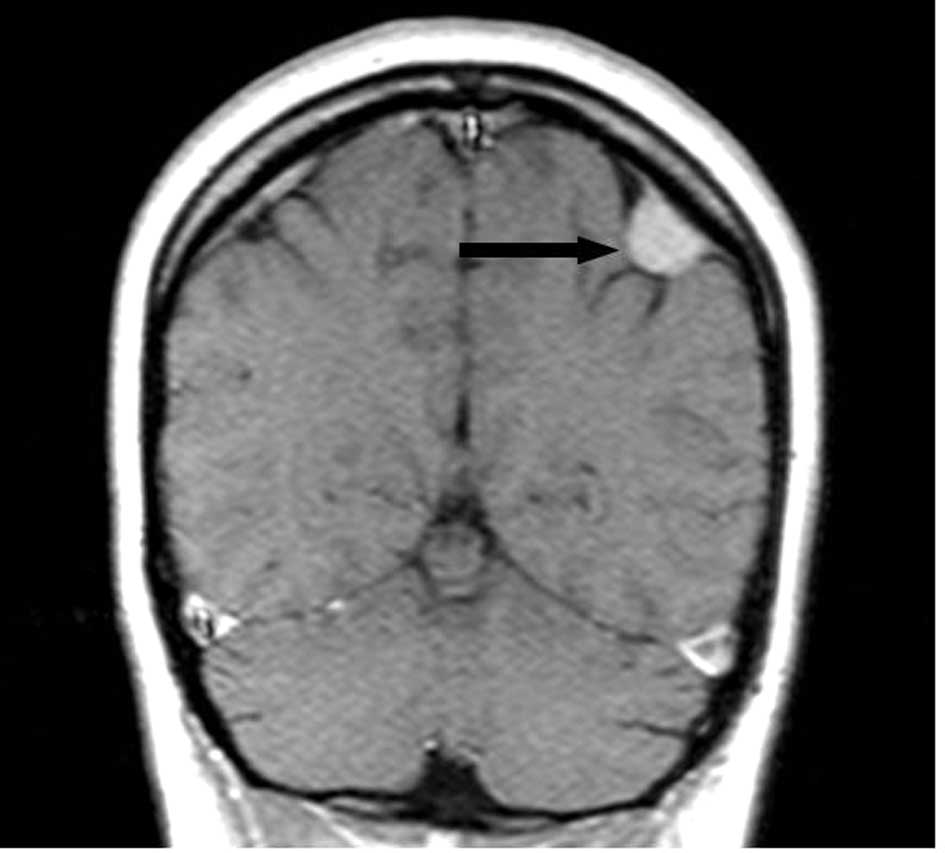
Screening for extrahepatic manifestation was
performed using magnetic resonance imaging (MRI), which revealed
the presence of a right frontal meningioma in the first patient
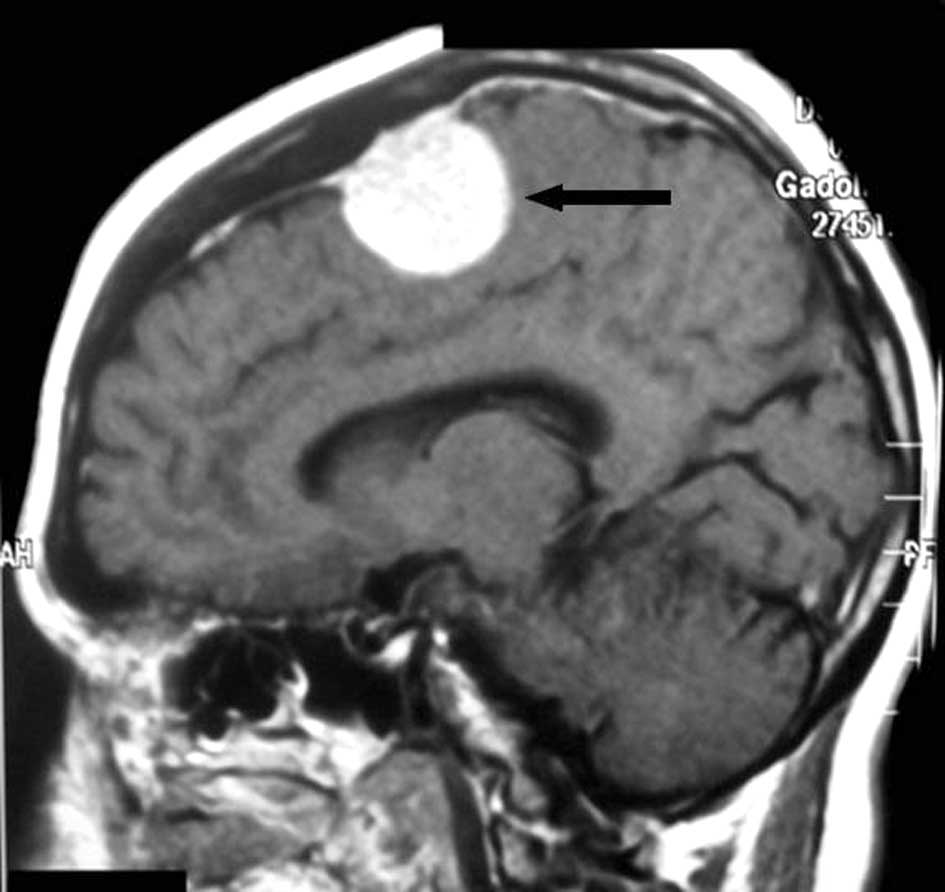
(Fig. 2), and a parietal posterior
calcified meningioma, plus the presence of osteoma and encephalic
calcification, in the second patient (Fig. 3), each measuring 1 and 7×3×2 cm in
diameter, respectively. The patients were submitted to a craniotomy
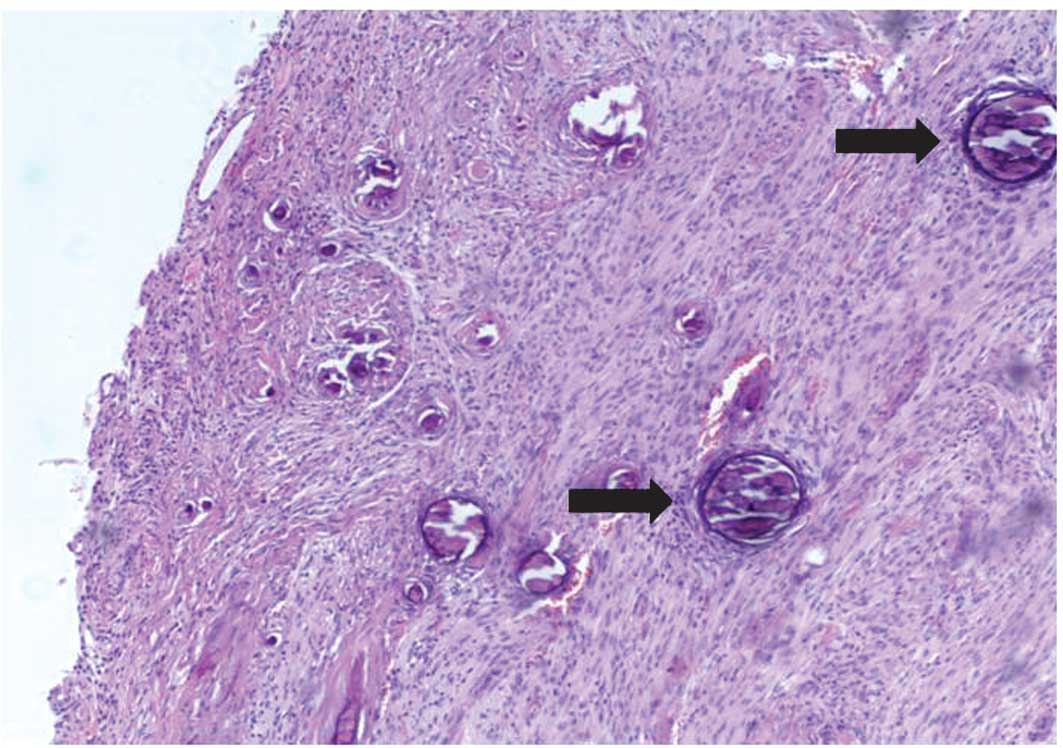
for tumor removal, with histological patterns compatible with
fibrous meningioma in the first patient, and transitional
meningioma in the second patient, both with psammomatous bodies
(Fig. 4). These histological
features lead to a classification of Grade I according to World
Health Organization (WHO) criteria. The postoperative course was
uneventful, with the patients still awaiting a liver
transplant.
Post-operative conventional cytogenetic
studies
Cells were obtained from the two patients and
cultivated for 24 or 48 h in MarrowMAXTM Bone Marrow
Medium (Gibco; Invitrogen, Brazil) with 10% fetal calf serum
without stimulation. The cells were then harvested using
conventional techniques. The GTG banding analysis was performed
according to Scheres (6). Metaphase
chromosomes were analyzed using an Olympus BX61 microscope
connected to BandView 4.5 software (Applied Spectral Imaging,
Israel). Chromosomes were identified and classified according to
the International System of Human Cytogenetic Nomenclature
guidelines (7). The analysis of
twenty metaphases for each patient did not reveal any cytogenetic
changes.
Discussion
The symptoms of isolated PCLD are specifically
correlated with the extension of liver involvement (8), with the possibility of
life-threatening complications and a poor quality of life.
No exact epidemiological data are currently
available regarding the incidence of PCLD in any population.
However, PCLD, without ADPKD, is a rare condition with a prevalence
of 0.05 or 0.13% in two autopsy series.
When associated with ADPKD, the prevalence reported
was 16% in one study and 93% in another (9,10). In
these cases, a relevant genetic defect was linked to the polycystic
kidney disease 1 (PKD1) locus on chromosome 16 (the main locus
responsible for the disease) (12)
and PKD2 locus on chromosome 4. No evidence of further locus
heterogeneity was described (11).
In a study conducted by Pirson et al
(12) of a family with PCLD not
associated with kidney cysts and transmitted through three
generations, linkage to mutations of the genetic markers of PKD1
and PKD2 were excluded. One report concluded that isolated PCLD
exists as a genetic disease, distinct from ADPKD1 and ADPKD2.
A third gene, protein kinase C substrate 80K-H
(PRKCSH), accounts for a comparatively rare, isolated form of
autosomal dominant PCLD, which displays no kidney involvement
(4). Waanders et al
(13) concluded that 16% of
patients with PCLD were found to have either PRKCSH or SEC63
mutations.
In a study of 20 individuals with PCLD, Yang et
al (14) reported that the
PRKCSH gene was not a major genetic cause, and that there may be at
least another locus responsible for the disease in Taiwanese
patients. Through analysis of eight Finnish families with PCLD,
Tahvanainen et al (15)
concluded that, in most families, PCLD is linked to a locus on
chromosome 19p13.2-13.1; however, the disease is genetically
heterogeneous with at least one more locus, which remains to be
found.
Associated morbidities were notably less frequent in
patients with PCLD when comparing individuals with PCLD linked to
ADPKD16, and included mitral valve prolapse, diverticuli, brain
aneurisms and intracranial arachnoid cysts (17).
Intracranial lesions in association with polycystic
diseases are typically incidental findings in routine
investigations, while symptomatic lesions may include headaches,
seizure or focal neurological deficits (18). However, the association of rare,
developmental, solid tumors in meningeal membrane (meningiomas)
with PCLD has yet to be reported.
The treatment of PCLD aims to symptomatically
relieve symptoms related to cysts, and includes conservative
approaches such as cyst decompression, liver resections or liver
transplantation. The main reason for the consideration of liver
transplantation in our patients was the large liver volumes,
leading to an abdominal compartmental syndrome.
The meningeal tumors were treated prior to liver
transplantation for obvious reasons, since the tumor measurement
following resection was 7×3×2 cm in our second patient.
Meningiomas are mostly benign tumors, classified as
Grade I, originating from the arachnoid cap cells and represent
13–26% of all intracranial tumors (19). They are more common in older age
individuals and in females. The role of hormones has yet to
beclarified, with a five-year survival of more than 80% in typical
meningiomas, but is poorer in atypical presentations. Papillary and
haemangiopericytic morphology, large tumors, high mitotic index,
absence of progesterone receptors, deletion and loss of
heterozygosis are all poor prognostic factors. Complete surgical
excision is the standard treatment. Radiotherapy may be used in the
clinical practice in atypical Grade II and malignant Grade III or
recurrent meningiomas (20).
The histological characterization of fibrous and
transitional meningiomas in the patients we studied lead to a
classification of Grade I according to WHO criteria (19).
The postoperative follow-up for the two patients was
uneventful, without complications, with the patients remaining on
the liver transplant waiting list.
Bearing in mind that the polymorphism is a feature
of extrahepatic lesions in PCLD, and is either associated, or not,
to ADPKD, improved awareness is mandatory in order to determine the
best approach for the treatment of these patients. The lack of a
genetic or familial association between these two cases shows they
are likely to have occurred by chance rather than represent a
previously unrecognized association between polycystic liver
disease and cranial meningioma.
The analysis of a large patient cohort may provide
us with more detailed information and a better understanding of the
genetic background of such occurrences.
At present, a more prudent course toward the
identification of PCLD remains a combination of refinement of the
genetic interval and continued gene identification based on the
genomic sequence of the region, with emphasis placed on the
understanding of liver cyst pathogenesis, which may lead to
non-surgical therapy for patients affected with symptomatic liver
disease (21).
Acknowledgements
All authors contributed to the study design and
approved the final version of the manuscript.
References
|
1
|
Schivink WI, Huston J, Torres VE and Marsh
WR: Intracranial cysts in autosomal dominant polycystic kidney
disease. J Neurosurg. 83:1004–1007. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Romao EA, Moyses Neto M, Teixeira SR,
Muglia VF, Vieira OM and Dantas M: Renal and extrarenal
manifestations of autosomal dominant polycystic kidney disease.
Braz J Med Biol Res. 39:533–538. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Masyuk T and LaRusso N: Polycystic liver
disease: new insights into disease pathogenesis. Hepatology.
43:906–908. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Everson GT, Taylor MRG and Doctor RB:
Polycystic disease of the liver. Hepatology. 40:774–782. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Portmann BC and Roberts EA: Developmental
abnormalities and liver disease in childhood. MacSweens Pathology
of the Liver. Burt AD, Portmann BC and Ferrell LD: Churchill
Livingstone Elsevier; China: pp. 174–176. 2007
|
|
6
|
Scheres JM: Identification of two
robertsonian translocations with a giemsa banding technique. Hum
Genet. 15:253–256. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mitelman F: ISCN: An International System
for Human Cytogenetic Nomenclature. Karger; Basel: pp. 72005
|
|
8
|
Shrestha R, Martina JA, Kerkhof R, et al:
Postmenopausal estrogen therapy selectively stimulates hepatic
enlargement in women with autosomal dominant polycystic kidney
disease. Hepatology. 26:1282–1286. 1997.
|
|
9
|
Karhunen PJ and Tenhu M: Adult polycystic
liver and kidney diseases are separate entities. Clin Genet.
30:29–37. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kwok MK and Lewin KJ: Massive hepatomegaly
in adult polycystic liver disease. Am J Surg Pathol. 12:321–324.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peters DJM, Spruit L, Saris JJ, Ravine D,
Sandkuijl LA, Fossdal R, Boersma J, et al: Chromosome 4
localization of a second gene for autosomal dominant polycystic
kidney disease. Nat Genet. 5:359–362. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pirson Y, Lannoy N, Peters D, Geubel A,
Gogot JF, Breuning M and Dumoulin CV: Isolated polycystic liver
disease as a distinct genetic disease, unlinked to polycystic
kidney disease 1 and polycystic disease 2. Hepatology. 23:249–252.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Waanders E, Morsche RH, Man RA, Jansen JB
and Drenth JP: Extensive mutational analysis of PRKCSH and SEC63
broadens the spectrum of polycystic liver disease. Hum Mutat.
27:830–836. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang AM, Shi SC, Chu CH, Wang TE and Yang
WS: PRKCSH genetic mutation was not found in taiwanese patients
with polycystic liver disease. Dig Dis Sci. 24:1–15. 2009.
|
|
15
|
Tahvanainen P, Tahvanainen E, Reijonen H,
Halme L, Kaariainen H and Hockerstedt K: Polycystic liver disease
is genetically heterogeneous: clinical and linkage studies in eight
Finnish families. J Hepatology. 38:39–43. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hoevenaren IA, Wester R, Schrier RW,
MacFann K, Doctor RB, Drenth JP and Everson GT: Polycystic liver:
clinical characteristics of patients with isolated polycystic liver
disease compared with patients with polycystic liver and autosomal
dominant polycystic kidney disease. Liver Int. 28:264–270. 2008.
View Article : Google Scholar
|
|
17
|
Leung GKT and Fan YW: Chronic subdural
haematoma and arachnoid cyst in autosomal dominant polycystic
kidney disease (ADPKD). J Clin Neurosci. 12:817–819. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Galassi E, Tognetti F, Gaist G, Fagioli L
and Frank G: CT scan and metrizamide CT cisternography in arachnoid
cysts of the middle cranial fossa: classification and
pathophysiological aspects. Surg neurol. 17:363–369. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Louis DN, Scheithauer BW, Budka H, Van
Deimling A and Kepes JJ: Meningiomas. Pathology and Genetics
Tumours of the Nervous System, WHO Classification of Tumours.
Kleihues P and Cavenee WK: Iaro press; Lyon: pp. 176–184. 2000
|
|
20
|
Marosi C, Hassler M, Roessler K, et al:
Meningioma. Crit Rev Oncol Hematol. 67:153–171. 2008. View Article : Google Scholar
|
|
21
|
Reynolds DM, Falk CT, Li A, King BF,
Kamath PS, Huston J, et al: Identification of a locus for autosomal
dominant polycystic liver disease, on chromosome 19p13.2–13.1. Am J
Hum Genet. 67:1598–1604. 2000.PubMed/NCBI
|