Introduction
Hepatocellular carcinoma (HCC) is diagnosed in more
than half a million individuals worldwide every year. Liver cancer
is the fifth most common cancer in males and the seventh most
common in females. Most of the burden of the disease (85%) is borne
in developing countries, with the highest incidence rates reported
in regions where infection with hepatitis B virus (HBV) is endemic,
including Southeast Asia and sub-Saharan Africa. Additional risk
factors for HCC are alcohol, toxins, inlcuding aflatoxin,
hemochromatosis, α1-antitrypsin deficiency and non-alcoholic fatty
liver disease (NAFLD). HCC rarely occurs before the age of 40 years
and reaches a peak at approximately 70 years of age. Rates of liver
cancer among males are two to four times as high as the rates among
females (1–5). Despite major efforts to improve the
diagnosis and treatment of HCC, therapeutic options remain limited.
Most patients, especially in Asia and sub-Saharan Africa, present
at end stages of the disease or with underlying liver cirrhosis and
consequently surgical options may no longer be indicated. Thus,
there is a need for novel therapeutic agents and strategies.
Despite its global significance, HCC is understudied compared with
other major lethal types of cancer, and hence, our knowledge of the
genomic alterations implicated in HCC initiation and progression is
fragmentary. An improved understanding of the molecular genetic
alterations specific to HCC may lead to the development of more
efficient methods of prevention, early diagnosis and cure of this
disease.
In this study, we carried out whole-exome sequencing
using DNA obtained from HCC and matched normal tissue and found six
previously unidentified variants in six genes. We further studied
the molecular effects of these variants.
Materials and methods
Tissue and DNA preparation
HCC tissue was obtained from a 69-year-old male
patient, who had chronic HBV infection and lymph node metastasis.
The matched normal tissue was obtained from a distance of 3–4 cm
from the HCC. Informed consent for the research was obtained from
the ethical committee of the Hospital. DNA of these tissues was
extracted using the traditional phenol chloroform method.
Informed consent was obtained from the patient. The
study was approved by the ethics committee of the First Affiliated
Hospital of Xinxiang Medical University, Weihui, China.
Targeted sequence capture and
sequencing
SeqCap EZ Human Exome Library version 2.0 (Roche
Diagnostics, Mannheim, Germany; 44.1 Mb regions are covered by the
probes. Cat no. 05860504001) was used for sequence capture
according to the manufacturer’s instructions. Genomic DNA (10 μg)
was used to prepare the library with the Truseq DNA Sample Prep kit
from Illumina (Illumina, San Diego, CA, USA; Cat. no. FC-121-1001).
Sequencing was performed at a concentration of 12 pM on an Illumina
Genome Analyzer IIx with paired-end 115-bp reads.
The genomic DNA library preparation, targeted
sequence capture and massively parallel sequencing were completed
by Guangzhou iGenomics Co., Ltd. (Guangzhou, China).
Alignment, variants calling and quality
control
The software BWA (version 0.5.9) (6) was used to align the paired-end reads
to the reference human genome (hg19). After the alignment, PCR
duplications were removed using the SAMtools software package
(version 0.1.16) (7). Candidate
somatic variants were identified with the VarScan 2 software
(version 2.2.8) and filtered by the accessory script (fpfilter.pl,
version 1.01) with default parameters (8). To qualify the identified somatic
variants, all candidate variants were subjected to manual review
using the Integrative Genomics Viewer (9). Common variants were excluded by
filtering known germ-line variants in Ensembl (version 64,
http://sep2011.archive.ensembl.org/index.html) and an
internal database composed of variants which occurred more than
twice in 40 publicly available control genomes. A variant was noted
if it was annotated as associated with a phenotype by Ensembl.
Bioinformatics analysis of non-synonymous
somatic variants
The effects of the non-synonymous somatic variants
were evaluated by bioinformatics analysis. The analysis of the
chemical polarity and conservation, prediction of secondary
structure and the domain of the proteins were performed. The
conservation analysis was carried out by PhyloP (10) and MutationTaster (11), the prediction of secondary structure
was performed through Jpred (12)
and the domain prediction was analyzed with UniProt (http://www.uniprot.org).
Results
Clinical information of the patient
A 69-year-old male patient was diagnosed with
pleomorphic cell-type HCC with lymph node metastasis. The primary
tumor was on the left lobe of the liver (10×8×6 cm in size) and
showed invasive and septal cirrhosis with macro- and micronodules.
It was a grade II to III HCC with prominent clear cell components.
The results of serological tests for HBV showed that the tumor was
positive for surface antigen, Anti-HBe (E) and core antibodies,
indicating that the patient had chronic HBV infection.
Summary of exome sequencing
The mean total number of bases sequenced was 4.65
Gb. The mean depth of the target region was 44-fold. The mean
percentage of the targeted bases which were covered at least once
was 98.6%. The mean percentage of the targeted bases which were
covered sufficiently for variant calling (coverage ≥5) was 92.1%.
The mean percentage of genes having >95% of their coding bases
called was 77.0%. The details of the quality are shown in Table I.
 | Table I.Summary of exome sequencing. |
Table I.
Summary of exome sequencing.
| Item | HCC | Matched normal | Average |
|---|
| Total bases
sequenced (Gb) | 4.64 | 4.66 | 4.65 |
| Mean depth of
target region (-fold) | 24 | 63 | 44 |
| Percentage of the
targeted bases which were covered at least once (%) | 98.4 | 98.8 | 98.6 |
| Percentage of the
targeted bases which were covered sufficiently for variant calling
(%) | 87.6 | 96.6 | 92.1 |
| Percentage of genes
having >95% of their coding bases called (%) | 70.4 | 83.5 | 77.0 |
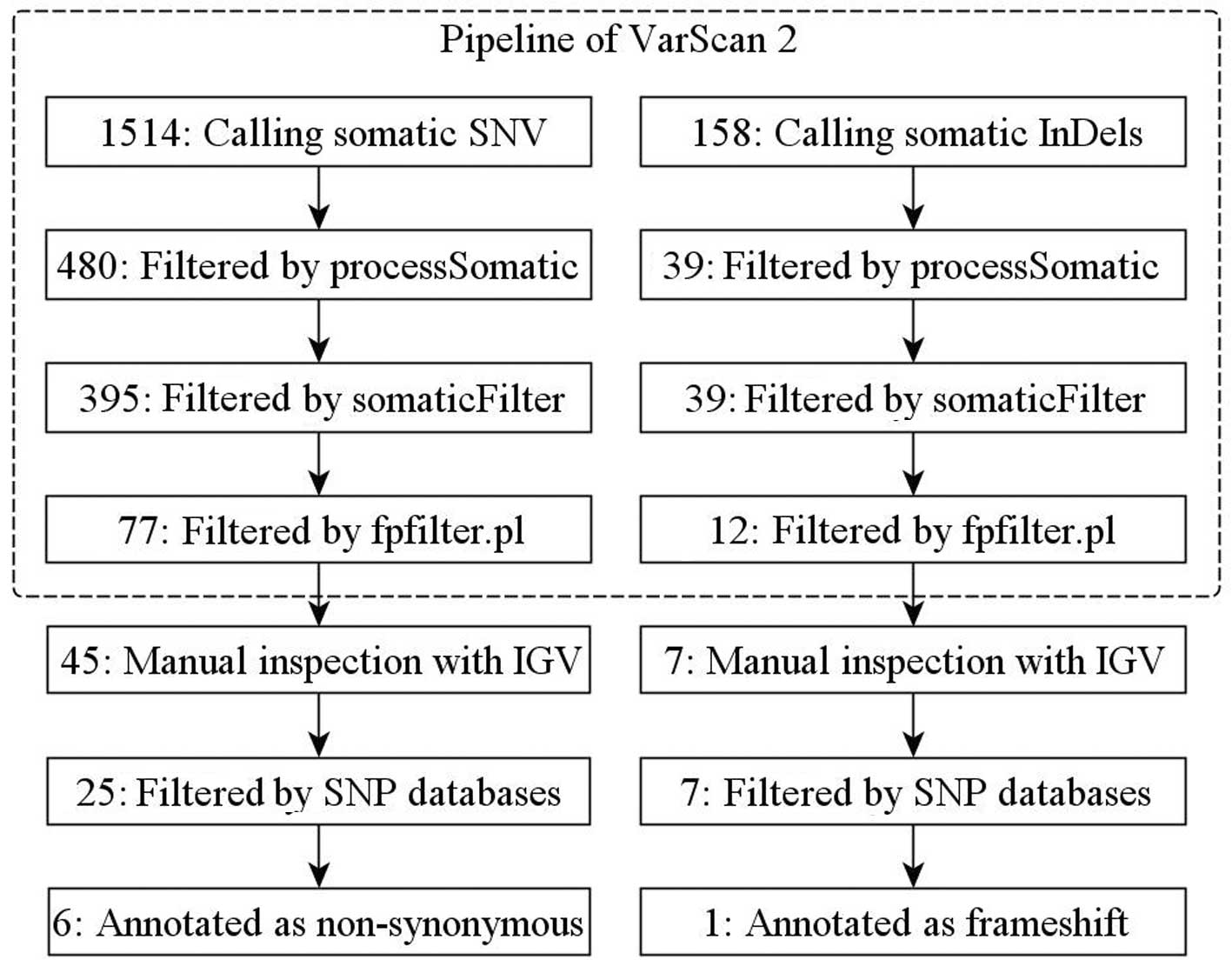
Discovery of somatic variants
Raw sequence data revealed 1514 candidate somatic
single nucleotide variations (SNVs) and 158 candidate somatic small
insertions and deletions (InDels) in the HCC tissue and 63047
candidate germ-line SNVs and 4513 InDels. A series of subsequent
qualifications of these data narrowed down these variants into 27
tumor-specific SNVs and seven InDels (Fig. 1). We focused our analysis on the six
non-synonymous substitutions and one frameshift mutation affecting
the integrity of the open reading frame (ORF). These seven
candidate variants were located in different genes, SPATA21,
PPCS, CDH12, OR1L3, PCK2, HUWE1
and PHF16. All the candidate variants were validated by PCR
and sequencing, with the exception of a non-synonymous substitution
in PPCS, which could not be distinguished from noise. Among
these genes, HUWE1 and PHF16 were in the X
chromosome. The details of the candidate variants are shown in
Table II.
| Table II.Non-synonymous somatic variants
identified in HCC by whole-exome sequencing. |
Table II.
Non-synonymous somatic variants
identified in HCC by whole-exome sequencing.
| Gene | Genomic
locusa | Accession no. | Mutation | Protein | Functionb |
|---|
| SPATA21 | Chr1: 16748433 | NM_198546 | c.C68T | p.T23M | Calcium ion
binding |
| PPCS | Chr1: 42922652 | NM_024664 | c.C416T | p.A139V | Phosphopantothenate
- cysteine ligase activity |
| CDH12 | Chr5: 22078699 | NM_004061 | c.G87C | p.Q29H | Calcium ion
binding |
| OR1L3 | Chr9:
125438256 | NM_001005234 | c.T848C | p.V283A | Odorant
receptor |
| PCK2 | Chr14:
24566276 | NM_001018073 | c.G205A | p.E69K | GTP binding/kinase
activity |
| HUWE1 | ChrX: 53596682 | NM_031407 | c.G6418A | p.A2140T | Acid - amino acid
ligase activity/binding |
| PHF16 | ChrX: 46887461 | NM_001077445 | c.642_643insA | p.G214fs | Zinc ion
binding |
Bioinformatics analysis of validated
variants
Six validated non-synonymous somatic variants were
involved in the bioinformatics analysis, including five
non-synonymous substitutions and one InDel. The chemical polarity
of all the variants was changed, with the exception of the
variation in OR1L3. Five variants would affect the secondary
structure of the protein and the variant of OR1L3 would not.
The frameshift variation in PHF16, located in the known
domain, disturbed the gene structure. The results of conservation
analysis indicated that the variants within PCK2 and
HUWE1 may disturb the function of the protein encoded by
these genes. The details of the analyzing of the candidate variants
are shown in Table III.
| Table III.Bioinformatics analysis of validated
variants. |
Table III.
Bioinformatics analysis of validated
variants.
| Gene | Mutation | Protein | Chemistry
polarity | Secondary structure
of protein | Domain | PhyloP value | Conservative
species at protein level |
|---|
| SPATA21 | c.C68T | P.T23M | Changed | Changed | Unknown | −2.074 | NA |
| CDH12 | c.G87C | p.Q29H | Changed | Changed | Unknown | 2.801 | Chimp, rhesus, cat,
mouse, chicken |
| OR1L3 | c.T848C | p.V283A | NA | NA | Known | 1.866 | Gorilla |
| PCK2 | c.G205A | p.E69K | Changed | Changed | Unknown | 5.208 | Rhesus, cat, mouse,
fugu, chicken, Xenopus, zebrafish, C. elegans,
Drosophila |
| HUWE1 | c.G6418A | p.A2140T | Changed | Changed | Unknown | 5.451 | Chimp, rhesus,
mouse, zebrafish, fugu |
| PHF16 | c.642_643insA | p.G214fs | Changed | Changed | Known | NA | NA |
Discussion
Using whole-exome sequencing, we identified seven
non-synonymous somatic variants and validated six variants, which
were previously unidentified in HCC. We further performed a
bioinformatics analysis of the validated variants. The results
suggest that the function of three mutated genes, PCK2,
HUWE1 and PHF16, may be markedly changed.
HCC is a major health problem worldwide (13). Despite its global significance,
liver cancer is understudied compared to other major lethal types
of cancer, and few known genetic influences on the deveopment of
HCC have been reported.
A number of studies in recent years have provided
evidence that the p53 tumor suppressor gene plays a major
role in hepatocarcinogenesis (14).
However, the frequency of p53 variants and its mutation
spectrum, with 75% missense variants, are exceptionally diverse in
their position and nature, affecting over 200 codons scattered
mainly throughout the central portion of the gene (15). There were no candidate somatic
variants found within p53 in the patient of the present
study.
The insulin-like growth factor (IGF) signaling
system is an essential regulator of growth and development
(16). The biological actions of
the axis comprise a complex network of molecules whose main
components are two high affinity mitogenic ligands: IGF1 and IGF2.
The type 1 IGF receptor (IGF1R) has tyrosine kinase activity, the
type 2 IGF receptor (IGF2R) is involved in the internalization and
degradation of IGF2 and at least six high-affinity IGF-binding
proteins (IGFBPs), which modulate the amount and bioactivity of
locally available IGFs. Despite its role in normal physiology, the
IGF axis is involved in the pathogenesis of several human
malignancies, including breast, colon, prostate, lung and liver
cancer (17). In HCC, the most
frequently described aberrant feature concerning this pathway is
overexpression of IGF2, which has been found in preneoplastic
lesions (18). This mitogen, highly
expressed during embryonic development, is markedly downregulated
after birth by tight epigenetic regulation of the P2–P4 fetal
promoters. Reactivation of IGF2 expression involves loss of
specific imprinting and hypomethylation (19). Allelic losses of IGF2R has been
detected in 60–70% of HCC cases, with inactivating variants in the
remaining allele also reported (20). In addition, reduced expression of
IGFBP-3 associated with promoter hypermethylation has been reported
in human HCC samples (21). A
recent study found aberrant activation of IGF1R in 21% of early
stage hepatitis C-related HCC xases, and provided preclinical
evidence of antineoplastic activity following IGF1R selective
blockade using a monoclonal antibody (22). The potential role of the HBx viral
protein as an inducer of IGF-IR expression has also been suggested
(23). In the present study, the
candidate gene PCK2 is in the insulin signaling pathway.
PCK2 encodes a member of the phosphoenolpyruvate
carboxykinase (GTP) family. The protein is a mitochondrial enzyme
that catalyzes the conversion of oxaloacetate to
phosphoenolpyruvate in the presence of GTP. A cytosolic form
encoded by a different gene has also been characterized and is the
key enzyme of gluconeogenesis in the liver. The encoded protein may
serve a similar function, although it is constitutively expressed
and not modulated by the hormones, including glucagon and insulin,
that regulate the cytosolic form. Hill et al reported that
the direct action of TNF to decrease the PEPCK transcription
rate was confirmed in vitro with H-4-II-E Reuber hepatoma
cells (24). The authors suggested
that PCK2 may be associated with hepatocarcinogenesis.
CTNNB1 and AXIN1 variants are
frequently found in HCC (25).
Variants prevent β-catenin ubiquitination and subsequent
degradation. Nuclear accumulation of β-catenin induces the
transcription of several genes associated with cell differentiation
and proliferation. In our study, the X-linked candidate gene
HUWE1 is in the ubiquitin-mediated proteolysis pathway. The
mutated gene may prevent β-catenin ubiquitination and subsequent
degradation, similar to CTNNB1 and AXIN1. Notably, it
is the X-linked gene which may contribute to the higher incidence
of HCC in males.
In conclusion, we identified six genes with
non-synonymous somatic variants in a HCC patient. Some of these
genes are involved in pathways associated with cell proliferation
and differentiation, which is known to be important in
hepatocarcinogenesis. Our results indicate that the insulin
signaling and ubiquitin-mediated proteolysis pathways may be
essential to HCC, and their relevant gene signature may be a target
for new therapies in HCC. Larger sample sizes are needed to confirm
or disprove our hypothesis.
Acknowledgements
We thank Dr Li Tong for the
suggestions about the experiment design. This study was supported
by the Provincial Education Science Foundation of Henan
(2009A330004).
References
|
1.
|
D Motola-KubaD Zamora-ValdésM UribeN
Méndez- SánchezHepatocellular carcinoma. An overviewAnn
Hepatol516242006
|
|
2.
|
P SrivatanakulH SriplungS
DeerasameeEpidemiology of liver cancer: an overviewAsian Pac J
Cancer Prev51181252004
|
|
3.
|
KA McGlynnWT LondonEpidemiology and
natural history of hepatocellular carcinomaBest Pract Res Clin
Gastroenterol19323200510.1016/j.bpg.2004.10.00415757802
|
|
4.
|
JM ClarkThe epidemiology of nonalcoholic
fatty liver disease in adultsJ Clin Gastroenterol40Suppl
1S5S10200616540768
|
|
5.
|
EK TeoKM FockHepatocellular carcinoma: an
Asian perspectiveDig Dis19263268200110.1159/00005069211935085
|
|
6.
|
H LiR DurbinFast and accurate long-read
alignment with Burrows-Wheeler
transformBioinformatics26589595201010.1093/bioinformatics/btp69820080505
|
|
7.
|
H LiB HandsakerA Wysoker1000 Genome
Project Data Processing Subgroup: The Sequence Alignment/Map format
and
SAMtoolsBioinformatics2520782079200910.1093/bioinformatics/btp35219505943
|
|
8.
|
DC KoboldtQ ZhangDE LarsonVarScan 2:
somatic mutation and copy number alteration discovery in cancer by
exome sequencingGenome
Res22568576201210.1101/gr.129684.11122300766
|
|
9.
|
JT RobinsonH ThorvaldsdóttirW
WincklerIntegrative genomics viewerNat
Biotechnol292426201110.1038/nbt.1754
|
|
10.
|
KS PollardMJ HubiszKR RosenbloomA
SiepelDetection of nonneutral substitution rates on mammalian
phylogeniesGenome Res20110121201010.1101/gr.097857.10919858363
|
|
11.
|
JM SchwarzC RödelspergerM SchuelkeD
SeelowMutationTaster evaluates disease-causing potential of
sequence alterationsNat
Methods7575576201010.1038/nmeth0810-57520676075
|
|
12.
|
C ColeJD BarberGJ BartonThe Jpred 3
secondary structure prediction serverNucleic Acids
Res36W197W201200810.1093/nar/gkn23818463136
|
|
13.
|
JM LlovetAM Di BisceglieJ BruixPanel of
Experts in HCC-Design Clinical Trials: Design and endpoints of
clinical trials in hepatocellular carcinomaJ Natl Cancer
Inst100698711200810.1093/jnci/djn134
|
|
14.
|
Y EdamotoA HaraW BiernatAlterations of
RB1, p53 and Wnt pathways in hepatocellular carcinomas associated
with hepatitis C, hepatitis B and alcoholic liver cirrhosisInt J
Cancer106334341200310.1002/ijc.11254
|
|
15.
|
P HainautM Hollsteinp53 and human cancer:
the first ten thousand mutationsAdv Cancer
Res7781137200010.1016/S0065-230X(08)60785-X10549356
|
|
16.
|
M PollakInsulin and insulin-like growth
factor signalling in neoplasiaNat Rev
Cancer8915928200810.1038/nrc253619029956
|
|
17.
|
D SachdevD YeeDisrupting insulin-like
growth factor signaling as a potential cancer therapyMol Cancer
Ther6112200710.1158/1535-7163.MCT-06-008017237261
|
|
18.
|
P Laurent-PuigJ Zucman-RossiGenetics of
hepatocellular
tumorsOncogene2537783786200610.1038/sj.onc.1209547
|
|
19.
|
SH TangDH YangW HuangHK ZhouXH LuG
YeHypomethylated P4 promoter induces expression of the insulin-like
growth factor-II gene in hepatocellular carcinoma in a Chinese
populationClin Cancer
Res1241714177200610.1158/1078-0432.CCR-05-226116857788
|
|
20.
|
AT De SouzaGR HankinsMK WashingtonTC
OrtonRL JirtleM6P/IGF2R gene is mutated in human hepatocellular
carcinomas with loss of heterozygosityNat
Genet1144744919957493029
|
|
21.
|
T HanafusaY YumotoK NousoReduced
expression of insulin-like growth factor binding protein-3 and its
promoter hypermethylation in human hepatocellular carcinomaCancer
Lett176149158200210.1016/S0304-3835(01)00736-4
|
|
22.
|
V TovarC AlsinetA VillanuevaIGF activation
in a molecular subclass of hepatocellular carcinoma and
pre-clinical efficacy of IGF-1R blockageJ
Hepatol52550559201010.1016/j.jhep.2010.01.01520206398
|
|
23.
|
SO KimJG ParkYI LeeIncreased expression of
the insulin-like growth factor I (IGF-I) receptor gene in
hepatocellular carcinoma cell lines: implications of IGF-I receptor
gene activation by hepatitis B virus X gene productCancer
Res563831383619968706031
|
|
24.
|
MR HillRE McCallumIdentification of tumor
necrosis factor as a transcriptional regulator of the
phosphoenolpyruvate carboxykinase gene following endotoxin
treatment of miceInfect Immun60404040501992
|
|
25.
|
A VillanuevaP NewellDY ChiangSL FriedmanJM
LlovetGenomics and signaling pathways in hepatocellular
carcinomaSemin Liver Dis275576200710.1055/s-2006-96017117295177
|