Introduction
Breast cancer is the most common neoplasia in women
and its pathogenesis is related to an acquired or inherited genetic
disorder influenced by environmental, behavioral or reproductive
factors (1). Cancer biomarker
discovery is important for both cancer biology and clinical
applications. These markers may come from DNA, RNA, miRNA or
proteins (2), with proteins being
the most significant (3).
The development and improvement of biotechnologies
has allowed researchers to perform high-throughput analyses of
genomes, transcriptomes and proteomes in health and disease, and
identify hundreds of potential biomarkers (4), offering the potential to discover
diagnostic, prognostic or therapeutic targets. However, less than
two dozen cancer biomarkers are currently approved by the Food and
Drug Administration (FDA) (5),
including only 9 protein biomarkers identified in the blood
(6). Due to the lack of sensitivity
and specificity of these known biomarkers (7), researchers continue to search for more
significant targets. Proteomics is a promising approach for the
discovery of cancer targets and biomarkers (8). The mapping of proteome profiles and
differential proteomics has been widely performed in breast cancer
to identify potential biomarkers (9). The identified proteins were reported
to have potential clinical significance, and certain proteins may
be used as potential diagnostic, prognostic or predictive
biomarkers (10,11,12,13,14).
However, due to the heterogeneity in the different studies,
including experimental design, sample collection and classification
and analytical method (15), these
results lack good reproducibility and require further validation
before they can be used in clinical detection and to explain the
underlying mechanisms of breast cancer. In addition, few protein
candidates were warranted to be specific to breast cancer, and were
often differentially expressed in other cancer types (16). Hence, research encountered the
challenge of how to decipher and use these individual results and
bring them into clinical applications. In addition, an
understanding of the underlying biological mechanisms of
carcinogenesis and the altered molecular events in breast cancer at
integrated pathway levels is necessary. Proteins do not act alone
to perform biological functions, but through complex biological
pathways. The discovery of these intricate pathways is essential to
understanding biological mechanisms. A wide variety of cancers may
also link to the same pathways that affect tumorigenesis and
progression through altering protein expressions. Therefore, once
these pathways are known, it may be easier to monitor different
aspects of cancer progression and develop a therapeutic strategy by
focusing on pathways instead of individual proteins. The enriched
pathways or functions may be the most probable cause of cancer
(17), and the enriched proteins
involved in these processes could in turn serve as target agents in
diagnosis or treatment. Several monoclonal antibodies and small
molecular inhibitors have been developed to target certain
molecular pathways involved in cell growth, survival and metastasis
in breast cancer (18,19,20).
Therefore, integrated bioinformatics should be applied when
discovering cancer-associated pathways and networks to improve our
understanding of cancer biology, as well as cancer diagnosis and
therapeutics.
In the present study, we integrated protein profiles
in breast cancers from literature and public databases to perform
bioinformatical analyses. Differential serum proteomics between
human breast cancer patients and healthy volunteers were performed,
and western blotting was used to validate the differential
expression of secretory proteins in breast cancer. Our aim was not
to discover all the breast cancer-associated proteins but rather to
focus on screening enriched signaling pathways in breast cancer
which may provide new insights into breast cancer research. This
bioinformatical insight into breast cancer-associated protein
profiles may potentially provide clues for identifying new
functional modules in breast cancer and may be used to understand
the underlying tumorigenesis process.
Materials and methods
Patient characteristics and serum
collection
Blood samples were collected from 25 breast cancer
patients and 20 healthy volunteers at the Yantai Yu-Huang-Ding
Hospital, China, with written informed consent and approval of the
Yu-Huang-Ding Hospital research and ethics committee. Venous blood
was drawn from each subject into 10-ml fasting blood tubes and
allowed to clot at room temperature for 1 h. Serum was separated by
centrifugation at 2000 g for 15 min at 4°C. Proteins in the
supernatant were precipitated by mixing with 4 volumes of ice-cold
acetone and allowing it to stand at −20°C for 1 h. After being
centrifuged at 12000 g for 1 h, washed with 90% acetone and dried,
the proteins were taken up in 2 ml lysis solution (7 M urea, 2 M
thiourea and 65 mM dithiothreitol) and stored at −80°C.
Collection of breast cancer-associated
proteins
Breast cancer protein profiles were constructed by
retrieving public databases (Uniprot, Release 2011_11, http://www.uniprot.org) and literature on differential
proteomes in breast cancer (dbDEPC 2.0, http://lifecenter.sgst.cn/dbdepc/index.do). All human
proteins in the Uniprot database were downloaded with all
annotations. Breast cancer-associated proteins were further
selected manually from the downloaded data using the keywords
‘cancer’, ‘tumor’ or ‘carcinoma’. The regulation levels were
recorded as ‘over-regulation’, ‘downregulation’ and
‘no-annotation’. All proteins extracted from the literature were
further grouped into over- and downregulation clusters, and certain
proteins with controversial expression in different reports were
clustered into the no-annotation group.
Bioinformatics analysis
Ontological analysis
All breast cancer-associated proteins were
classified into different protein classes according to Interpro
(www.ebi.ac.uk/interpro) and Gene Ontology (GO)
annotation (www.geneontology.org), and significantly
enriched functions were further selected according to Panther
(http://www.pantherdb.org/).
Pathway analysis
Ingenuity Pathway Analysis v8.0-2803 (Ingenuity
Systems, Redwood City, CA, USA) was used to analyze pathways and
networks involving the breast cancer-associated proteins. The
following settings were used: reference set, Ingenuity Knowledge
Base (genes only); network analysis, direct and indirect
relationships; molecules per network, 35; networks per analysis,
25. All species, tissues and cell lines were used for the analysis.
IPA uses Fisher’s exact test to determine which pathways (canonical
pathways, toxicity pathways or biological functions) are
significantly linked to the input protein set compared with the
whole Ingenuity Knowledge Base.
Selection of secretory and cell
surface proteins
Secretory proteins and cell surface proteins are
promising biomarkers. All cancer-associated proteins were compared
with the serum/plasma proteome (21) to select secretory proteins, and
compared with the cell surfaceome (22) to select cell surface proteins. GO
was used to further filter the results.
Proteomic analysis of human serum from
patients with breast cancer and healthy volunteers
Two-dimensional gel electrophoresis and mass
spectrometric analyses were performed as described in our previous
study (23). Gels were made in
triplicate to confirm the spot patterns and were scanned with a
Z320 scanner (Founder, Beijing, China). The gel images were
processed with ImageMaster software (GE Healthcare, Piscataway, NJ,
USA). Images were briefly checked manually to eliminate artefacts.
Following spot detection, a match set was built including all the
experimental and control gels. The significance of the differential
expression of protein spots between experimental and control groups
was estimated by mean ratio >2.0 and the independent samples
t-test; P<0.05 was considered to indicate a statistically
significant result.
Western blot analysis
A total of 50 μg of pooled proteins were separated
at 12% SDS-PAGE by electrophoresis and then transferred onto a
nitrocellulose membrane (Millipore, Bedford, MA, USA). The
membranes were blocked with 5% milk TBST (10 mM Tris-HCl, pH 8.0,
150 mM NaCl, 0.1% Tween-20) overnight at 4°C and then hybridized
with the following primary antibodies: anti-ORM2 (sc-51020),
anti-APOA1 (sc-69755), anti-GC (sc-18706) and anti-CLU (sc-166907),
all from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
immune complexes were visualized with a DAB staining kit (Zhongshan
Jinqiao Technology, China).
Results
Overview of breast cancer-associated
proteins
The breast cancer-associated protein profile was
mainly comprised of proteins screened from the Uniprot database and
literature reports. A total of 803 breast cancer-associated
proteins were screened from literature reports and 243 were
selected in the Uniprot database. Finally, 1031 breast
cancer-associated proteins were obtained including 514 (49.6%)
upregulated and 318 (30.8%) downregulated proteins. One hundred and
seventy-two proteins required further confirmation of
expression.
Bioinformatics analysis
From protein lists to biological
functions
According to GO analysis, Interpro and literature
annotations, the enriched biological functions are listed in
Table I. Metabolic process,
chaperone transcription and catalytic activity-associated proteins
were more significantly enriched in breast cancer-associated
proteins. The comparison of up- and downregulated proteins
demonstrated that more chaperone, cell adhesion, transporter and
antioxidant-related proteins were present in the upregulated group,
while defense/immunity and extracellular proteins were present in
the downregulated group.
 | Table IBroad biological functions of breast
cancer protein profiles. |
Table I
Broad biological functions of breast
cancer protein profiles.
| Protein profiles
a
| Upregulated
proteins
| Downregulated
proteins
|
|---|
| Biological
functions | Observed nos. | Expected nos. | P-value | Observed nos. | Expected nos. | P-value | Observed nos. | Expected nos. | P-value |
|---|
| Antioxidant
activity | 4 | 0.75 |
7.10e−3 | 10 | 1.41 |
2.31e−6 | 3 | 0.43 |
9.28e−3 |
| Apoptosis | 36 | 24.84 |
1.81e−2 | 73 | 46.96 |
1.78e−4 | 24 | 14.17 |
8.92e−3 |
| Binding | 158 | 173.60 |
7.86e−2 | 358 | 328.21 |
2.39e−2 | 126 | 99.01 |
6.36e−4 |
| Calmodulin | 7 | 1.60 |
1.34e−3 | 1 | 0.85 |
5.72e−1 | 1 | 0.48 |
3.84e−1 |
| Catalytic
activity | 213 | 137.21 |
3.29e−13 | 375 | 259.42 |
4.35e−16 | 104 | 78.25 |
5.80e−4 |
| Cell adhesion | 13 | 4.52 |
8.11e−4 | 12 | 2.39 |
7.51e−6 | 0 | 1.36 |
2.55e−1 |
| Cell-cell
signaling | 19 | 34.23 |
2.61e−3 | 43 | 64.71 |
2.04e−3 | 15 | 19.52 |
1.74e−1 |
| Chaperone | 28 | 6.32 |
1.56e−10 | 22 | 3.34 |
8.96e−12 | 5 | 1.91 |
4.40e−2 |
| Cytoskeletal
protein | 55 | 21.44 |
5.64e−10 | 27 | 11.34 |
4.24e−5 | 16 | 6.47 |
9.47e−4 |
| Defense/immunity
protein | 10 | 5.20 |
3.93e−2 | 2 | 2.75 |
4.81e−1 | 8 | 1.57 |
2.15e−4 |
| Extracellular
matrix protein | 7 | 3.50 |
6.50e−2 | 1 | 1.85 |
4.47e−1 | 5 | 1.06 |
4.50e−3 |
| G-protein coupled
receptor | 1 | 21.73 |
6.62e−9 | 0 | 11.49 |
8.94e−6 | 0 | 6.56 |
1.32e−3 |
| Hsp70 family
chaperone | 5 | 0.63 |
4.94e−4 | 4 | 0.33 |
3.95e−4 | 1 | 0.19 |
1.74e−1 |
| Isomerase
activity | 12 | 4.86 |
4.19e−3 | 26 | 9.19 |
3.65e−6 | 6 | 2.77 |
6.18e−2 |
| Lyase activity | 17 | 5.30 |
3.54e−5 | 24 | 10.01 |
1.11e−4 | 6 | 3.02 |
8.50e−2 |
| Metabolic
process | 283 | 212.58 |
2.58e−10 | 529 | 401.91 |
1.39e−16 | 157 | 121.24 |
1.64e−5 |
|
Metalloprotease | 16 | 7.05 |
2.47e−3 | 7 | 3.73 |
8.36e−2 | 4 | 2.13 |
1.66e−1 |
| Oxidoreductase
activity | 59 | 18.08 |
3.86e−15 | 105 | 34.18 |
1.40e−23 | 31 | 10.31 |
7.17e−8 |
| Peptidase
activity | 28 | 18.44 |
2.06e−2 | 53 | 34.86 |
2.10e−3 | 14 | 10.51 |
1.72e−1 |
| Peptidase inhibitor
activity | 11 | 4.55 |
6.96e−3 | 22 | 8.61 |
8.61e−5 | 6 | 2.60 |
4.79e−2 |
| Peroxidase
activity | 4 | 0.69 |
5.55e−3 | 8 | 1.31 |
6.73e−5 | 2 | 0.40 |
6.04e−2 |
| Protease | 42 | 23.14 |
2.23e−4 | 22 | 12.24 |
6.84e−3 | 11 | 6.98 |
9.46e−2 |
| Protease
inhibitor | 15 | 5.30 |
3.93e−4 | 6 | 2.80 |
6.48e−2 | 5 | 1.60 |
2.32e−2 |
| Protein
transport | 67 | 42.33 |
1.43e−4 | 111 | 80.02 |
3.43e−4 | 23 | 24.14 |
4.58e−1 |
| Receptor
activity | 30 | 46.49 |
4.84e−3 | 56 | 87.90 |
9.95e−5 | 19 | 26.51 |
7.17e−2 |
| Structural molecule
activity | 62 | 38.26 |
1.39e−4 | 133 | 72.34 |
1.34e−11 | 48 | 21.82 |
2.42e−7 |
| Transcription
factor | 44 | 100.49 |
3.02e−11 | 20 | 53.15 |
5.28e−8 | 17 | 30.31 |
4.39e−3 |
| Transfer/carrier
protein | 29 | 12.06 |
2.12e−5 | 10 | 6.38 |
1.11e−1 | 7 | 3.64 |
7.51e−2 |
| Transferase
activity | 59 | 40.96 |
3.27e−3 | 96 | 77.45 |
1.85e−2 | 25 | 23.36 |
3.92e−1 |
| Translation
factor | 10 | 2.72 |
5.21e−4 | 6 | 1.44 |
3.61e−3 | 3 | 0.82 |
5.03e−2 |
| Transport | 99 | 73.47 |
1.18e−3 | 183 | 138.90 |
5.80e−5 | 50 | 41.90 |
1.04e−1 |
| Vesicle coat
protein | 6 | 1.46 |
3.86e−3 | 6 | 0.77 |
1.48e−4 | 0 | 0.44 |
6.44e−1 |
| Unclassified | 93 | 171.80 |
6.09e−15 | 95 | 173.91 |
7.31e−15 | 45 | 97.98 |
2.04e−12 |
From biological functions to
pathways
To explore the enrichment pathways of these proteins
with different biological functions, a pathway analysis was
performed using Ingenuity Pathway Analysis tools and the Kyoto
Encyclopedia of Genes and Genomes (KEGG) database. Thirty-three
pathways were identified in breast cancers, including 25 pathways
enriched in upregulated proteins and 8 pathways with downregulated
proteins (Table II).
| Table IICanonical pathways in up- and
downregulated breast cancer proteins. |
Table II
Canonical pathways in up- and
downregulated breast cancer proteins.
| Pathway | Ratio | Number | P-value |
|---|
| Upregulated | | | |
| Glycolysis | 0.18 | 15 |
4.88e−8 |
| Pyruvate
metabolism | 0.18 | 11 |
3.52e−6 |
| Protein
ubiquitination pathway | 0.09 | 23 |
1.30e−5 |
| RhoA
signaling | 0.12 | 13 |
5.00e−5 |
| NRF2-mediated
oxidative stress response | 0.09 | 17 |
6.71e−5 |
| PI3K/AKT
signaling | 0.10 | 13 |
8.75e−5 |
| Fatty acid
metabolism | 0.13 | 13 |
2.62e−4 |
| Integrin
signaling | 0.08 | 16 |
2.80e−4 |
| ILK
signaling | 0.08 | 15 |
2.82e−4 |
| 14-3-3-mediated
signaling | 0.10 | 12 |
3.10e−4 |
| RAN
signaling | 0.28 | 5 |
3.16e−4 |
| Aryl hydrocarbon
receptor signaling | 0.12 | 17 |
3.48e−4 |
| Clathrin-mediated
endocytosis signaling | 0.08 | 14 |
6.40e−4 |
| Valine, leucine
and isoleucine degradation | 0.13 | 8 |
7.71e−4 |
| IGF-1
signaling | 0.10 | 10 |
1.12e−3 |
| VEGF
signaling | 0.10 | 9 |
1.75e−3 |
| Arginine and
proline metabolism | 0.11 | 8 |
2.10e−3 |
| EIF2
signaling | 0.07 | 14 |
2.19e−3 |
| Actin
cytoskeleton signaling | 0.07 | 15 |
2.96e−3 |
| ERK5
signaling | 0.11 | 7 |
3.55e−3 |
| GABA receptor
signaling | 0.13 | 6 |
4.00e−3 |
| Regulation of
actin-based motility by Rho | 0.09 | 8 |
5.16e−3 |
| LPS/IL-1 mediated
inhibition of RXR function | 0.06 | 13 |
9.42e−3 |
| HER-2 signaling
in breast cancer | 0.09 | 7 |
1.10e−2 |
| Downregulated | | | |
| Citrate
cycle | 0.09 | 5 |
1.25e−4 |
| Acute phase
response signaling | 0.06 | 10 |
6.53e−4 |
| P53
signaling | 0.07 | 7 |
1.13e−3 |
| Primary
immunodeficiency signaling | 0.09 | 5 |
1.80e−3 |
| Urea cycle and
metabolism of amino groups | 0.13 | 4 |
2.76e−3 |
| Cdc42
signaling | 0.06 | 8 |
3.40e−3 |
| Glyoxylate and
dicarboxylate metabolism | 0.18 | 4 |
1.12e−3 |
| Autoimmune
thyroid disease signaling | 0.08 | 3 |
1.02e−3 |
From biological pathways to
networks
Network generation was performed using Ingenuity
Pathway Analysis tools. This generated 25 networks using all the
breast cancer-associated proteins and 13 networks using the
upregulated proteins. Several functions were linked to more than 3
of the networks in the upregulated group and the main ones included
cancer, genetic disorder, cell death and cellular movement.
Cancer/testis proteins, secretory
proteins and cell surface proteins
Cancer/testis antigens are potential cancer
biomarkers with restricted expression in the testis and certain
cancer cells. The earlier comparison of breast cancer proteins
showed that 119 testis proteins including 11 testis-specific
proteins were included in this study. Twenty-one proteins were
common to both the breast cancer proteins and cancer/testis
antigens. The comparison of breast cancer proteins with human
serum/plasma and cell surface proteins (surfaceome) revealed that
the cancer proteins included 153 secretory proteins and 69 cell
surface proteins
Experimental analysis by
2D-electrophoresis and western blotting
All gels were statistically analyzed through
ImageMaster software, and 11 proteins were shown to be
differentially expressed between breast cancer patients and healthy
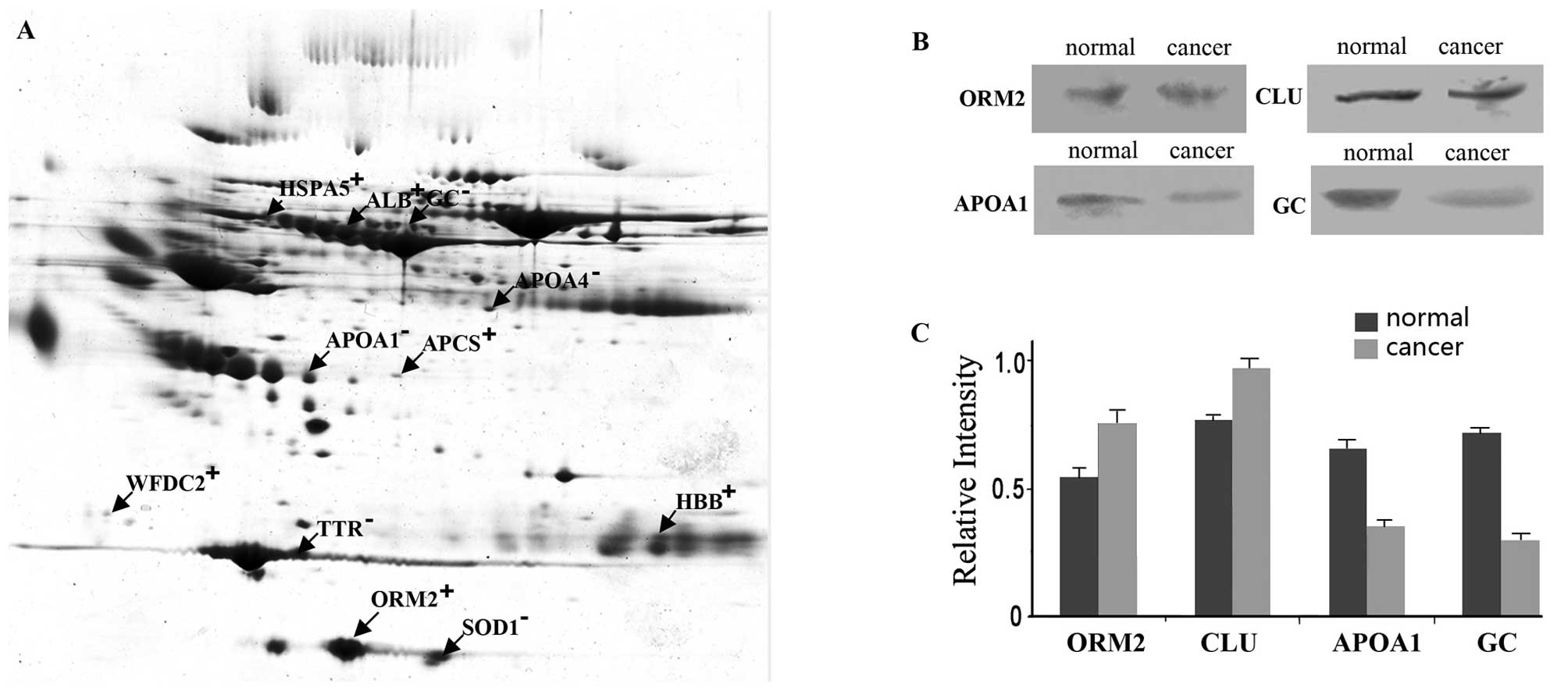
donors (Fig. 1A). Of the 4 proteins
tested by western blotting, ORM2 and CLU were verified to be highly
expressed in the serum of breast cancer patients, and APOA1 and GC
were expressed at low levels (Fig.
1B).
Discussion
With the development and improvement of proteomic
technology, cancer proteomes are evolving quickly and aid in the
search for biomarkers and therapeutic targets. Breast cancer
proteomes were widely analyzed and certain important proteins were
identified (9,10). However, these data were
heterogeneous and many of them lacked experimental verification and
validation. In our preliminary study, 11 serum proteins were found
to be differentially expressed between breast cancer patients and
healthy individuals. The results provided insight into certain
aspects of breast cancer, but had little correlation with all the
breast cancer proteomes available. Thus, the integration of protein
lists containing breast cancer proteomes to understand the
correlations involved in regulation functions and networks may aid
in the screening of diagnostic markers or therapeutic targets.
In the present study, integrated bioinformatical
tools were used to analyze the enriched pathways and networks that
are associated with breast cancer. The breast cancer protein
profile was constructed, including proteins in the published
literature of differential proteomics and the Uniprot database.
Protein expressions in breast cancer were evaluated, and broadly
categorized into three groups: upregulated, downregulated and those
that had contradictory expressions in breast cancer. An ontological
analysis indicated that most of these breast cancer-associated
proteins were classified into different enrichment functional
groups that may be involved in different aspects of cancer biology.
The pathway networks analysis showed that several signaling
pathways and networks were significantly associated with human
breast cancer. These pathways and functions were relevant to the
cancer microenvironment, invasion and metastasis processes as
briefly described below.
Previous studies have shown that the cancer
microenvironment is deficient in oxygen, low in glucose (24), and is is usually followed by highl
glycolytic activity (25,26). Studies of breast cancers showed that
hypoxic conditions may alter the expression of certain proteins
that serve as hypoxic markers (27). Fifteen glycolytic enzymes were
investigated in the present study which may be involved in tumor
proliferation in hypoxic conditions. The protein ubiquitination
pathway and the PI3K/Akt signaling pathway have been reported to be
associated with the development of human breast cancers (28,29).
Certain drugs have been applied to affect these pathways in the
treatment of breast cancer (30,31,32,33).
The RhoA signaling pathway was strongly correlated with tumor cell
invasion and metastasis (34), and
the Nrf2-mediated oxidative stress response activated genes
encoding detoxification enzymes and antioxidant proteins to protect
cells from oxidative stress. In breast cancer cells, NRP/B was able
to enhance oxidative stress responses via the Nrf2 pathway
(35). More than 13 cell adhesion
proteins in the present study may play roles in the intercellular
and cell-extracellular matrix interactions of cancer, leading to
cancer invasion or metastasis (36), or participating in signal
transduction, cell growth and differentiation (37). E-cadherin is one prominent adhesion
molecule that forms the E-cadherin-catenin complex which plays a
role in epithelial cell-cell adhesion and differentiation (38), in particular serving as a potent
invasion/tumor suppressor of breast cancer (39). Previous studies indicated that the
downregulation of E-cadherin was relevant to several pathways
including the integrin-linked kinase (ILK) signaling pathway
(40,41). The ILK signaling pathway was one
prominent enrichment pathway in breast cancer, and may play an
important role in hormonal cancer progression (42).
Some of these canonical pathways are known to
participate in cancer processes, but the mechanism and the altered
individual members are not well studied. We hypothesized that the
altered expressions of different members or activators in these
enriched pathways may be associated with the tumor process and its
microenvironment alteration, and may serve as important targets or
agents in the diagnosis and treatment of breast cancer.
More than 200 cancer/testis antigens have been
listed in the cancer/testis database (http://www.cta.lncc.br/). The significant feature of
these proteins is their restricted expression in the testis and low
or no expression in normal tissues (43); therefore, they may be used as
potential cancer vaccine targets. Twenty-one cancer/testis antigens
(8%) and 119 testis proteins (11 testis-specific proteins) were
included in the breast cancer protein profiles and these proteins
warrant further study. Among these proteins, a recent study showed
that MAGE-A3 and A4 in the peripheral blood of breast cancer
patients may have potential prognostic and predictive implications
(44). All of these proteins are
promising specific tumor markers of breast cancer.
Due to the limitations of protein identification
using current proteomic techniques, some molecules in certain
pathways may be missing. However, the present study gives a new
bioinformatical insight into breast cancer at systems biology
levels by integrating the individual studies to identify enriched
biological and molecular pathways, providing vital evidence for
future research. Further studies are warranted to substantiate the
enriched functions and pathways. The studies may advance our
understanding of cancer biomarker discovery, and also facilitate
the biological interpretation of cancer biology in a network
context.
References
|
1.
|
E WarnerClinical practice. Breast-cancer
screeningN Engl J
Med36510251032201110.1056/NEJMcp110154021916640
|
|
2.
|
C AggarwalN SomaiahGR SimonBiomarkers with
predictive and prognostic function in non-small cell lung cancer:
ready for prime time?J Natl Compr Canc Netw8822832201020679541
|
|
3.
|
Y TanSY MaFQ WangHP MengC MeiA LiuHR
WuProteomic-based analysis for identification of potential serum
biomarkers in gallbladder cancerOncol Rep26853859201121687958
|
|
4.
|
MS Abu-AsabM ChaouchiS AlesciS GalliM
LaassriAK CheemaF AtoufJ VanMeterH AmriBiomarkers in the age of
omics: time for a systems biology
approachOMICS15105112201110.1089/omi.2010.002321319991
|
|
5.
|
BK DunnPD WagnerD AndersonP
GreenwaldMolecular markers for early detectionSemin
Oncol37224242201010.1053/j.seminoncol.2010.05.00720709207
|
|
6.
|
JA LudwigJN WeinsteinBiomarkers in cancer
staging, prognosis and treatment selectionNat Rev
Cancer5845856200510.1038/nrc173916239904
|
|
7.
|
II WistubaJG GelovaniJJ JacobySE DavisRS
HerbstMethodological and practical challenges for personalized
cancer therapiesNat Rev Clin
Oncol8135141201110.1038/nrclinonc.2011.221364686
|
|
8.
|
S Schmitz-SpankeAW RettenmeierProtein
expression profiling in chemical carcinogenesis: a proteomic-based
approachProteomics11644656201110.1002/pmic.20100040321246732
|
|
9.
|
TY LauDP O’ConnorDJ BrennanMJ DuffySR
PenningtonWM GallagherBreast cancer proteomics: clinical
perspectivesExpert Opin Biol
Ther7209219200710.1517/14712598.7.2.20917250459
|
|
10.
|
H HondermarckC TastetI El
Yazidi-BelkouraRA ToillonX Le BourhisProteomics of breast cancer:
the quest for markers and therapeutic targetsJ Proteome
Res714031411200810.1021/pr700870c18311906
|
|
11.
|
D BöhmK KellerN WehrweinA LebrechtM
SchmidtH KölblFH GrusSerum proteome profiling of primary breast
cancer indicates a specific biomarker profileOncol
Rep2610511056201121837365
|
|
12.
|
M KadowakiT SangaiT NagashimaM SakakibaraH
YoshitomiS TakanoK SogawaH UmemuraK FushimiY NakataniF NomuraM
MiyazakiIdentification of vitronectin as a novel serum marker for
early breast cancer detection using a new proteomic approachJ
Cancer Res Clin
Oncol13711051115201110.1007/s00432-010-0974-921253761
|
|
13.
|
B BalluffM ElsnerA KowarschS RauserS
MedingC SchuhmacherM FeithK HerrmannC RöckenRM SchmidH HöflerA
WalchMP EbertClassification of HER2/neu status in gastric cancer
using a breast-cancer derived proteome classifierJ Proteome
Res963176322201010.1021/pr100573s21058730
|
|
14.
|
TC LaiHC ChouYW ChenTR LeeHT ChanHH ShenWT
LeeST LinYC LuCL WuHL ChanSecretomic and proteomic analysis of
potential breast cancer markers by two-dimensional differential gel
electrophoresisJ Proteome
Res913021322201010.1021/pr900825t20052998
|
|
15.
|
HJ IssaqTJ WaybrightTD VeenstraCancer
biomarker discovery: Opportunities and pitfalls in analytical
methodsElectrophoresis32967975201110.1002/elps.20100058821449066
|
|
16.
|
B WeigeltL PusztaiA AshworthJS
Reis-FilhoChallenges translating breast cancer gene signatures into
the clinicNat Rev Clin
Oncol95864201110.1038/nrclinonc.2011.12521878891
|
|
17.
|
CL SawyersThe cancer biomarker
problemNature452548552200810.1038/nature0691318385728
|
|
18.
|
CM SchlotterU VogtH AllgayerB
BrandtMolecular targeted therapies for breast cancer
treatmentBreast Cancer Res10211200810.1186/bcr211218671839
|
|
19.
|
H MukaiTargeted therapy in breast cancer:
current status and future directionsJpn J Clin
Oncol40711716201010.1093/jjco/hyq03720382634
|
|
20.
|
C Bernard-MartyF LebrunA AwadaMJ
PiccartMonoclonal antibody-based targeted therapy in breast cancer:
current status and future
directionsDrugs6615771591200610.2165/00003495-200666120-0000416956305
|
|
21.
|
SJ LiM PengH LiBS LiuC WangJR WuYX LiR
ZengSys-BodyFluid, a systematical database for human body fluid
proteome researchNucleic Acids
Res37D907D912200910.1093/nar/gkn84918978022
|
|
22.
|
JP da CunhaPA GalanteJE de SouzaRF de
SouzaPM CarvalhoDT OharaRP MouraSM Oba-ShinjaSK MarieWA Silva
JrBioinformatics construction of the human cell surfaceomeProc Natl
Acad Sci1061675216757200919805368
|
|
23.
|
J LiF LiuH WangX LiuJ LiuN LiF WanW WangC
ZhangS JinJ LiuP ZhuY LiuSystematic mapping and functional analysis
of a family of human epididymal secretory sperm-located proteinsMol
Cell Proteomics925172528201010.1074/mcp.M110.00171920736409
|
|
24.
|
HF DvorakVM WeaverTD TlstyG BergersTumor
microenvironment and progressionJ Surg
Oncol103468474201110.1002/jso.2170921480238
|
|
25.
|
RA GatenbyRJ GilliesWhy do cancers have
high aerobic glycolysis?Nat Rev
Cancer4891899200410.1038/nrc147815516961
|
|
26.
|
WH KoppenolPL BoundsCV DangOtto Warburg’s
contributions to current concepts of cancer metabolismNat Rev
Cancer113253372011
|
|
27.
|
H BandoM ToiK KitadaM KoikeGenes commonly
upregulated by hypoxia in human breast cancer cells MCF-7 and
MDA-MB-231Biomed
Pharmacother57333340200310.1016/S0753-3322(03)00098-214568227
|
|
28.
|
M OsakiM OshimuraH ItoPI3K-Akt pathway:
its functions and alterations in human
cancerApoptosis9667676200410.1023/B:APPT.0000045801.15585.dd15505410
|
|
29.
|
SE GhayadPA CohenInhibitors of the
PI3K/Akt/mTOR pathway: new hope for breast cancer patientsRecent
Pat Anticancer Drug
Discov52957201010.2174/15748921078970220819751211
|
|
30.
|
RZ OrlowskiEC DeesThe role of the
ubiquitination-proteasome pathway in breast cancer: applying drugs
that affect the ubiquitin-proteasome pathway to the therapy of
breast cancerBreast Cancer Res517200310.1186/bcr46012559038
|
|
31.
|
S RossiM LodaThe role of the
ubiquitination-proteasome pathway in breast cancer: use of mouse
models for analyzing ubiquitination processesBreast Cancer
Res51622200310.1186/bcr54212559040
|
|
32.
|
S LipkowitzThe role of the
ubiquitination-proteasome pathway in breast cancer: ubiquitin
mediated degradation of growth factor receptors in the pathogenesis
and treatment of cancerBreast Cancer
Res5815200310.1186/bcr54112559039
|
|
33.
|
PF McAuliffeF Meric-BernstamGB MillsAM
Gonzalez-AnguloDeciphering the role of PI3K/Akt/mTOR pathway in
breast cancer biology and pathogenesisClin Breast
Cancer10S59S65201010.3816/CBC.2010.s.01321115423
|
|
34.
|
AP StruckhoffMK RanaRA WorthylakeRhoA can
lead the way in tumor cell invasion and metastasisFront
Biosci1619151926201110.2741/383021196273
|
|
35.
|
S SengHK AvrahamS JiangS YangM SekineN
KimelmanH LiS AvrahamThe nuclear matrix protein, NRP/B, enhances
Nrf2-mediated oxidative stress responses in breast cancer
cellsCancer
Res6785968604200710.1158/0008-5472.CAN-06-378517875699
|
|
36.
|
TA MartinMD MasonWG JiangTight junctions
in cancer metastasisFront Biosci16898936200110.2741/3726
|
|
37.
|
SH KimJ TurnbullS GuimondExtracellular
matrix and cell signalling: the dynamic cooperation of integrin,
proteoglycan and growth factor receptorJ
Endocrinol209139151201110.1530/JOE-10-037721307119
|
|
38.
|
BP WijnhovenWN DinjensM
PignatelliE-cadherincatenin cell-cell adhesion complex and human
cancerBr J
Surg879921005200010.1046/j.1365-2168.2000.01513.x10931041
|
|
39.
|
G BerxF Van RoyThe E-cadherin/catenin
complex: an important gatekeeper in breast cancer tumorigenesis and
malignant progressionBreast Cancer
Res3289293200110.1186/bcr30911597316
|
|
40.
|
C TanP CostelloJ SangheraD DominguezJ
BaulidaAG de HerrerosS DedharInhibition of integrin linked kinase
(ILK) suppresses beta-catenin-Lef/Tcf-dependent transcription and
expression of the E-cadherin repressor, snail, in APC−/− human
colon carcinoma cellsOncogene20133140200111244511
|
|
41.
|
V BravouG KlironomosE PapadakiS TaravirasJ
VarakisILK over-expression in human colon cancer progression
correlates with activation of beta-catenin, down-regulation of
E-cadherin and activation of the Akt-FKHR pathwayJ
Pathol2089199200610.1002/path.186016278819
|
|
42.
|
V CortezBC NairD ChakravartyRK
VadlamudiIntegrin-linked kinase 1: role in hormonal cancer
progressionFront Biosci (Schol
Ed)3788796201110.2741/s18721196412
|
|
43.
|
A GrigoriadisOL CaballeroKS HoekL da
SilvaYT ChenSJ ShinAA JungbluthLD MillerD CloustonJ CebonLJ OldSR
LakhaniAJ SimpsonAM NevilleCT-X antigen expression in human breast
cancerProc Natl Acad
Sci1061349313498200910.1073/pnas.090684010619651608
|
|
44.
|
YM HusseinAF GharibRL EtewaAS El-ShalME
Abdel-GhanyWH ElsawyThe melanoma-associated antigen-A3, -A4 genes:
relation to the risk and clinicopathological parameters in breast
cancer patientsMol Cell
Biochem351261268201110.1007/s11010-011-0734-421264495
|