Introduction
Endometrial carcinoma is commonly believed to have a
relatively good prognosis. Hysterectomy with bilateral oophorectomy
usually leads to complete remission and long-term survival,
although surgery deprives these patients of fertility potential
(1,2). However, approximately 2–14% of
patients who suffer from endometrial carcinoma are below aged 40.
The younger patients are frequently nulligravid with a history of
infertility and a strong desire to preserve fertility (1–5).
Therefore, a more conservative medical treatment is desirable in
young patients who wish to preserve their fertility. Progestins,
particularly medroxyprogesterone acetate (MPA), have long been used
as a conservative treatment for young patients with clinical stage
I, grade I endometrial carcinoma. The response rate varies from
57–75% (1–3). It has been reported that conservative
treatment followed by IVF enables such patients to achieve a
successful pregnancy. These relatively good response rates are
encouraging with respect to the possibility of conservative
treatment by progestins in young patients (1–5).
However, more than 30% of patients with endometrial adenocarcinoma
displayed resistance to endocrine therapies at time of
presentation. Additionally, most cancer patients that initially
respond to progestin treatment will at some point develop
resistance, resulting in tumor progression (2). Progestin therapy is limited by the
development of resistance in the tumor. The cellular mechanisms
underlying acquired resistance to progestin are poorly understood.
In order to investigate the molecular mechanisms whereby human
endometrial adenocarcinoma develops resistance to progestin
therapy, we have undertaken to develop human endometrial
adenocarcinoma cell lines that are resistant to the
growth-inhibitory effects of progestins in vitro. Continuous
culture of breast cancer cell lines in the presence of antiestrogen
has led to the development of a number of resistant subcell lines
(6–8). These represent potentially important
models for study of the loss of endocrine sensitivity and the
acquisition of the resistant phenotype. In this study, we developed
endometrial carcinoma progestin-resistant subcell lines by similar
methods. The data presented in this study describe the
characteristics of a progestin-resistant subcell line developed
from Ishikawa human endometrial adenocarcinoma cells by stepwise
selection in increasing concentrations of the synthetic progestin,
MPA. The study was approved by the ethics committee of Zhengzhou
university, Zhengzhou, China.
Materials and methods
Routine cell culture and establishment of
progestin-resistant cell line
Ishikawa human endometrial carcinoma cells were
maintained in our laboratory. These were obtained from an
endometrial adenocarcinoma of a 39-year-old woman in 1985 by
Nishida, and established as ER- and PR-positive. The cells were
routinely cultured in DMEM/F12 medium (Gibco Life Technologies,
Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (Gibco
Life Technologies) at 37°C in a humidified atmosphere of 5%
CO2. Expressions of ER and PR in parent Ishikawa cells
were confirmed by immunocytochemistry. To generate
progestin-resistant subcell lines of Ishikawa cells, cells were
maintained in the above media supplemented with MPA (Sigma, St.
Louis, MA, USA) with 2.5 μM increases in MPA concentration
(1.0–10 μM) every 4 weeks. When the surviving cells had
grown to a high density but were still less than confluent, cells
were subcultured using 0.02% EDTA and 0.25% trysine prepared in
Hanks’ balanced salt solution. 1/50 of total cells were passaged.
Medium containing MPA was replenished every 2–3 days. At each
dosage level, several aliquots of cells were frozen and stored in
liquid nitrogen. Cells proliferating in 10 μM MPA with the
same doubling time as the parent Ishikawa cells proliferating in
the medium without MPA, were designated progestin-resistant
Ishikawa cells (6).The cells were
thereafter maintained in 10 μM MPA. All treatment stocks
were initially prepared in DMSO (vehicle) with subsequent dilution
for experiments of more than 1:1000 (for 10−5 M). The
presence of a vehicle at such dilutions has previously been shown
to have no effect on cell growth.
MTT assay
In some studies, cell number was determined by a MTT
assay. MTT (thiazolyl blue) is converted from a yellow-colored salt
to a purple-colored formazan by cleavage of the tetrazolium ring by
mitochondrial dehydrogenases, the activity of which is linear with
cell number. Cells were plated in a 96-well flat-bottomed
microplate in 100 μl culture medium per well at a cell
density of 1–2×104 cells/ml. After treatment as
indicated, 10 μl MTT (5 mg/ml in PBS) solution was added to
each well. After incubation for 4 h, the medium was poured off, and
formazan crystals were dissolved with 150 μl DMSO by
shaking. The absorbance was measured at 490 nm with a microplate
reader. Eight wells were used for each treatment and experiments
were repeated at least three times.
Flow cytometry
The parent Ishikawa cells and the
progestin-resistant Ishikawa cells were plated at a density of
1.5×106 per 6-cm-diameter dish (Corning, Inc., Corning,
NY, USA) and allowed to grow for 24 h. Subsequently, the medium was
changed to serum-free medium and 16 h later, the cells were
incubated with experimental medium. The hormone control group
received DMSO (vehicle) alone, whereas the hormone treatment group
received 1 μM MPA daily. Each experiment was performed in
triplicate. Cells were harvested 1, 2, 3 and 4 days after treatment
by trypsinization, washed twice with PBS and pelleted by
centrifugation for 5 min at 500 × g. Following this, they were
resuspended in PBS and fixed with 75% cold ethanol. Cells were then
resuspended in 1 ml of PI (Sigma) solution containing propidium
iodide 50 μg, and 100 units RNase. Cells were analyzed in a
flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). DNA
histograms were prepared using the ModFit analysis program (BD
Biosciences), which provides fits for the G0/G1, S and G2/M
fractions of the population. The S- and G2/M-phase fractions were
combined into a single growth fraction, the proliferative
index.
Matrigel invasion assays
The invasion assay of tumor cells was performed
using a Transwell cell culture chamber (Corning Costar No. 3422;
Corning, Inc.). Prior to the invasion experiments, cells were
cultured in serum-free medium containing 1 μM MPA or DMSO
(vehicle) for 72 h. Polycarbonate filters (10 mm) were coated on
the upper surface with Matrigel (10 mg/200 ml; BD Biosciences, San
Jose ,CA, USA). After the filters were dried at room temperature,
they were washed gently with phosphate-buffered saline (PBS). Cells
were harvested with 0.25% trypsin, washed and resuspended in
serum-free DMEM/F12. The upper compartment of the Matrigel invasion
chamber was loaded with 2×105 cells, and the lower
compartment was filled with 600 μl 1% FBS in DMEM/F12 to act
as an attractant. The plate was incubated at 37°C for 24 h. The
cells on the upper side of the filters were gently wiped off; the
filters were fixed in methanol and stained with hematoxylin and
eosin. The cells that had migrated to the lower side of the filters
were counted under a light microscope. The numbers of cells in five
defined, high-power fields (magnification, ×200) were counted and
the average was determined.
Immunocytochemical analysis
The parent Ishikawa cells and the
progestin-resistant Ishikawa cells were trypsinized, washed twice
with PBS and resuspended in 200 μl PBS. Subsequently, 5
μl cell suspension was smeared on the slides, dried in air,
then fixed by incubation at room temperature for 30 min with 3.7%
formaldehyde-phosphate-buffered saline and then washed twice with
PBS. The cell slides were then immunocytochemically stained, as
follows, and endogenous peroxidase activity was blocked by using 3%
hydrogen peroxide. The slides were microwaved in 10 mM citrate
buffer (pH 6.0) to unmask the epitopes. After incubation with
blocking solution for 15 min at room temperature, the slides were
incubated overnight at 4°C with monoclonal antibody against ERα
(1:100; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), ERβ
(1:200; Upstate Biotechnology, Inc., Lake Placid, NY, USA), PR
(1:100; Neomarker, Inc., Fremont, CA, USA) and PRB (1:100;
Neomarker). Following two 5-min PBS washes, the slides were
incubated with biotin-labeled secondary antibody for 30 min. Then,
after two 5-min PBS washes, ABC solution (a streptavidin-biotin
horseradish complex) was added for 15 min followed by two PBS
washes. The slides were then stained by exposure for 2 min to
diaminobenzidine and 0.04% H2O2. They were
then counterstained with hematoxylin, dehydrated and mounted.
Breast carcinoma cell line MCF-7 was used as a positive control.
Negative controls were made by replacing the primary antibody with
mouse serum. Negative controls yielded non-specific membrane
staining. Staining indices were determined by two independent
observers.
Statistical analysis
All calculations were performed using the SPSS
software package 11.0, ANOVA assay and paired t-test. A P-value
less than 0.05 was considered to indicate a statistically
significant difference.
Results
Establishment of the progestin-resistant
Ishikawa cell and its growth characteristics in MPA
It has been shown that Ishikawa human endometrial
cancer cell line is sensitive to the growth-inhibitory effects of
MPA in culture. The cells which are resistant to the
growth-inhibitory effects of MPA were selected from the parent
Ishikawa cells by stepwise selection in increasing concentrations
of MPA (from 1–10 μM). The cells were thereafter (passage
23) routinely maintained with 10 μM MPA in their culture
medium. Under this regimen, dramatically slowed growth rates were
observed for approximately 24 weeks from initial MPA exposure,
after which time cell growth rates progressively increased. The
progestin-resistant Ishikawa cells were achieved over a period of
approximately 10 months. The doubling time of the
progestin-resistant Ishikawa cells (34.18±3.15 h) grown routinely
in the medium containing 10 μM MPA, was not significantly
different to the doubling time of the parent Ishikawa cells
(35.14±2.68 h) grown in the absence of MPA (t=−0.331, P=0.762).
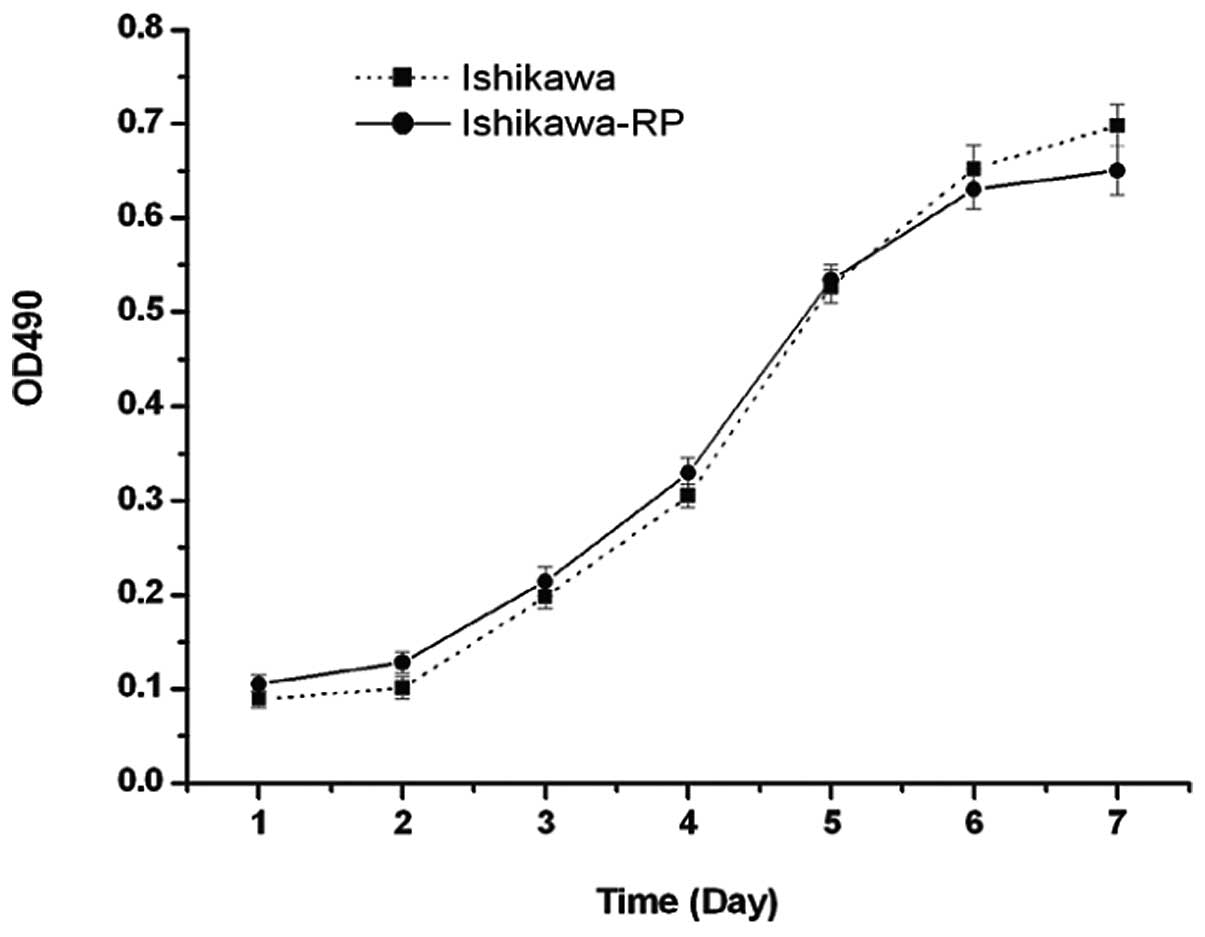
Fig. 1 shows the comparison of the
parent Ishikawa cells and the progestin-resistant Ishikawa cells
growth curves in their routine culture medium.
Effects of MPA on the growth of the
parent Ishikawa cells and the progestin-resistant Ishikawa
cells
MTT assay was used to determine the effects of MPA
on the growth of the parent Ishikawa cells and the
progestin-resistant Ishikawa cells. Treatment with MPA reduced the
growth of the parent Ishikawa cells. The inhibitory effect was
dose-dependent (F=29.525, P=0.000); the inhibitory effects were 20,
28 and 41%, respectively, when 0.1, 1 and 10 μM of MPA were
added. Notably, we found that the effect of treatment with MPA
shifted from growth suppression, as observed in the parent Ishikawa
cells, to growth stimulation in the progestin-resistant Ishikawa
cells (F=36.20, P=0.00). Low concentration of MPA (0.01 and 0.1
μM) had no significant effect on the growth of the
progestin-resistant cells; the stimulatory effects were 21 and 46%
respectively when 1 and 10 μM of MPA were added (Fig. 2).
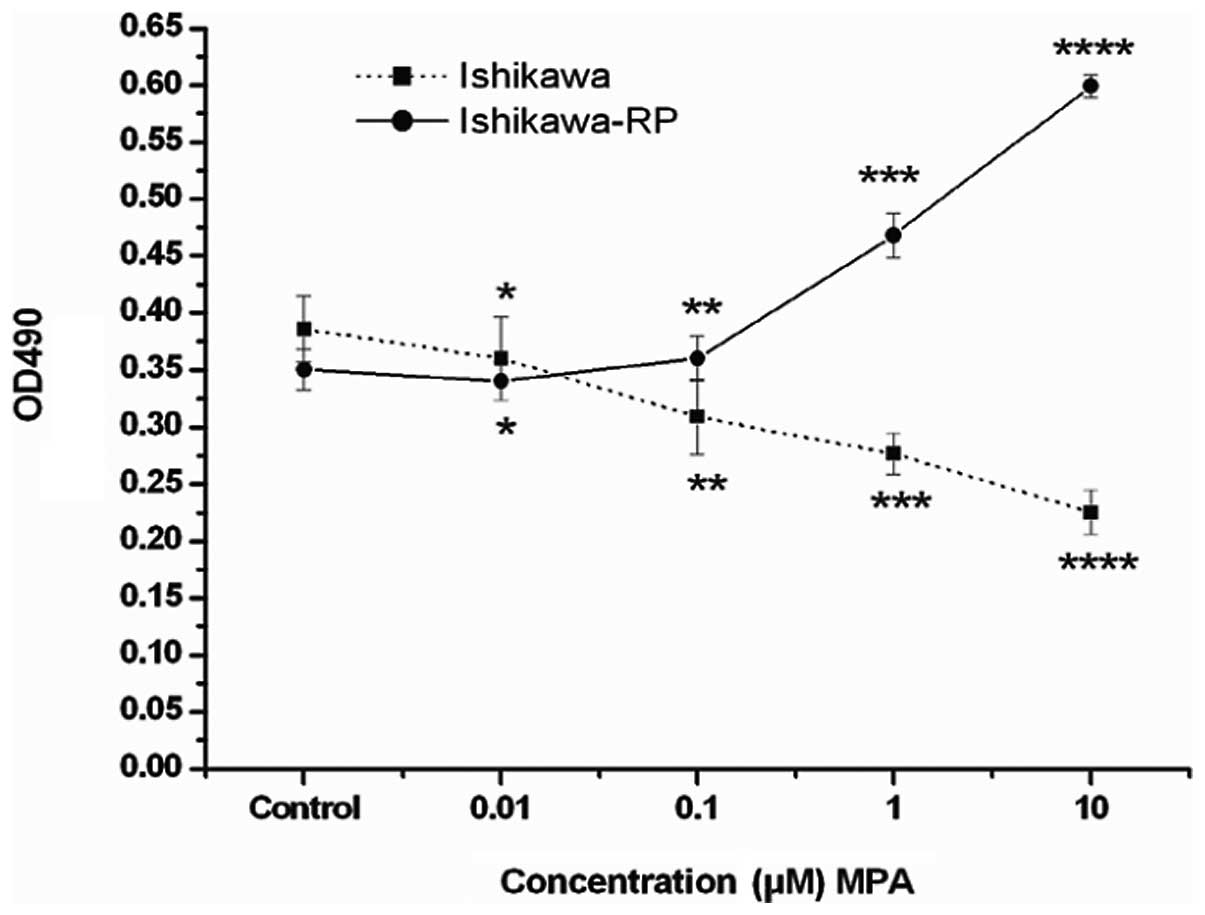 | Figure 2Effects of increasing concentrations
of MPA on the proliferation of the parent Ishikawa cells and the
progestin-resistant Ishikawa cells. MTT assay was used. Cells were
plated in 96-well flat-bottomed plates and allowed to grow for 24
h, then the medium was changed to serum-free medium. After 16-18 h
serum starvation, the cells were incubated with experimental medium
containing various concentrations of MPA (0.01, 0.1, 1 and 10
μM) for 72 h. The control group received DMSO (vehicle)
alone. MPA inhibited the parent Ishikawa cell growth. The
inhibitory effect was concentration-dependent. ANOVA analysis:
F=29.525, P=0.000, compared with control. *P=0.130,
**P=0.0000, *** P=0.0000,
****P=0.000. MPA stimulates progestin-resistant Ishikawa
cell proliferation, a relatively high concentration of MPA exerts
significant stimulatory effect. ANOVA analysis: F=30.369, P=0.000,
compared with control. *P=0.188, **P=0.333,
***P=0.001 and ****P=0.000. Bars, standard
error. MPA, medroxyprogesterone acetate. |
Effects of MPA on cell cycle distribution
in the parent Ishikawa cells and the progestin-resistant Ishikawa
cells
Flow cytometry was performed to determine the
fraction of cells in the presence of MPA in the parent Ishikawa
cells and the progestin-resistant cells. MPA caused time-dependent
inhibition of the cell cycle of the parent Ishikawa cells; MPA
produced a 14.2% fall in the proliferation index at day 1 and a 45%
reduction by day 4 (Fig. 3A). In
contrast, MPA had no effect on the cell cycle of the
progestin-resistant Ishikawa cells on days 1 and 2, but by days 3
and 4, MPA produced an increase in the proliferation index (21.9
and 20.85%, respectively; Fig.
3B).
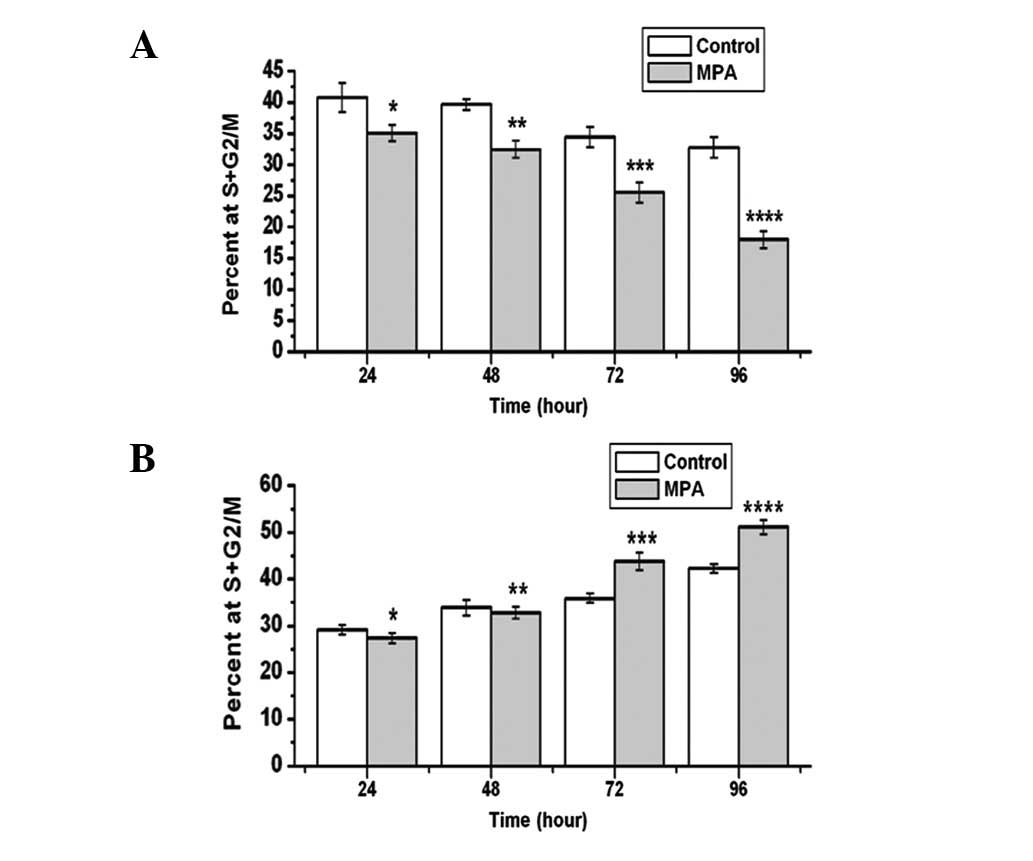 | Figure 3Effects of MPA on the cell cycle of
the parent Ishikawa cells and the progestin-resistant Ishikawa
cells. DMSO (control) or 1 μM MPA was added and the
experimental medium refreshed every day. Cells were harvested 1, 2,
3 and 4 days after treatment. The percentage of cells in S and G2/M
was measured by flow cytometry. Bars, standard error. (A) MPA
inhibited the parent Ishikawa cell proliferation, compared with
control,*t=3.539, P=0.035; **t=8.114,
P=0.002; ***t=4.429, P=0.011; ****t=5.964,
P=0.004; (B) MPA stimulated the progestin-resistant Ishikawa cell
proliferation, compared with control: *t=2.062, P=0.108;
**t=1.206, P=0.344; ***t=10.505, P=0.001;
****t=7.496, P=0.002. MPA, medroxyprogesterone
acetate. |
Effects of MPA on the invasion capability
of the parent Ishikawa cells and the progestin-resistant Ishikawa
cells
To assess the effects of MPA on cell invasiveness
and metastatic potential, cells were seeded on a Matrigel invasion
chamber after treatment with MPA or vehicle alone. Cells that
successfully invaded through the Matrigel and the pores on the
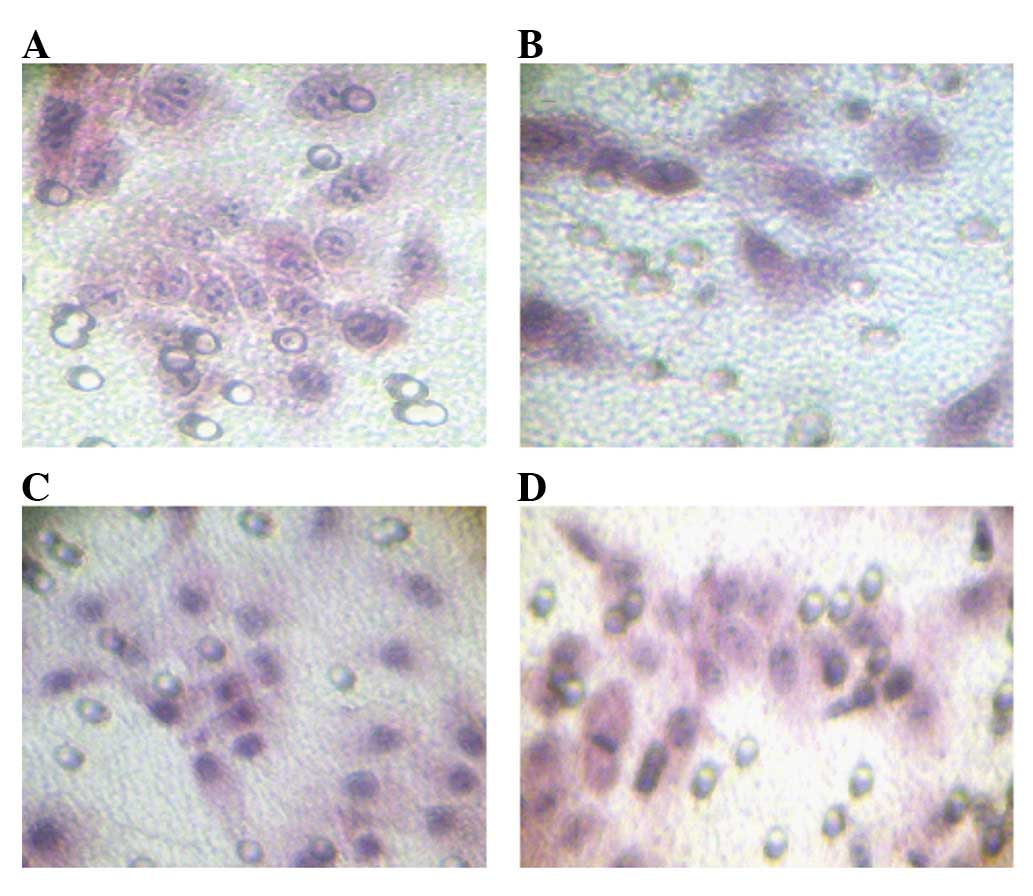
filter were stained with hematoxylin and eosin (Fig. 4) The invasive capability of the
progestin-resistant Ishikawa cells was higher than that of the
original Ishikawa cells. Notably, a dramatic inhibition of
invasiveness caused by MPA treatment in Ishikawa cells (t=6.107,
P=0.026) and the promotion of invasiveness caused by MPA treatment
in progestin-resistant Ishikawa cells (t=8.660, P=0.013) were
observed.
Comparison of steroid receptor expression
between the parent Ishikawa cells and the progestin-resistant
Ishikawa cells
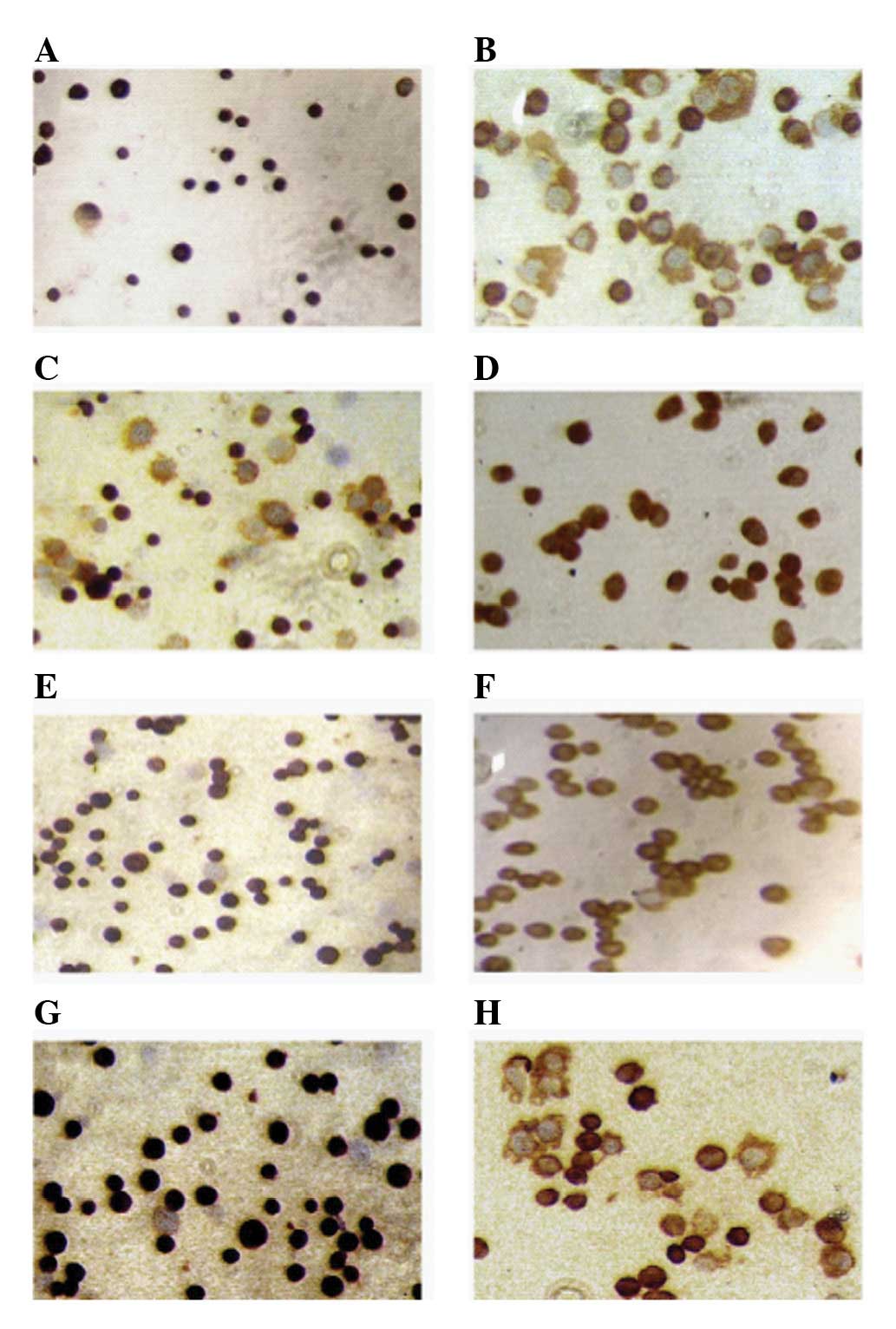
The positive rates of ERα, PR and PRB were over 95%
and the positive rate of ERβ was 56±5% in the parent Ishikawa
cells. Compared with parent Ishikawa cells, the positive rates of
ERα and PRB in the progestin-resistant Ishikawa cells were reduced
to 28±3% and 40±5% respectively, and the positive rate of ERβ was
statistically enhanced to 93.6±4.5%. These differences were
significant (P<0.05). The positive rate of PR in the
progestin-resistant Ishikawa cells was not different from the
parent Ishikawa cells, but the staining intensity was reduced. It
could be inferred that downregulation of the B subtype of PR may be
due to decreased PR staining intensity. The A subtype of PR was
maintained constantly, as shown in Fig.
5.
Discussion
In human endometrial cancer, the emergence of
progesterone-resistant cells reduces the efficacy of progesterone
therapy. In order to understand some of the possible mechanisms by
which hormonally dependent endometrial cancers develop resistance
to progestin therapy, we have developed an endometrial cancer cell
line which is resistant to the growth-inhibitory effects of the
synthetic progestin, MPA. The progestin-resistant Ishikawa cells,
developed from Ishikawa human endometrial cancer cells by stepwise
elevation of MPA concentration over a period of 10 months, were
grown routinely in the presence of 10 μM MPA with a doubling
time not significantly different from the parent Ishikawa cell line
grown in the absence of progesterone. The parent Ishikawa cells
were sensitive to the inhibitory effect of MPA; MPA treatment
showed growth suppression in a dose- and time-dependent fashion.
This is consistent with previous reports (1,10). In
contrast to the parent Ishikawa cells, the progestin-resistant
Ishikawa cells were resistant to the growth-inhibitory effects of
MPA and inversely, MPA exerted a growth-stimulatory effect. This is
in accordance with ‘M’ cells, a cell line selected for resistance
to the growth-inhibitory effects of MPA from MCF-7 cells, where MPA
appears to exert a growth-stimulatory effect (7). It is also concordant with an
antiestrogen-resistant cell line selected from an MCF-7 cell line
(8). On the other hand, MPA not
only exerted a growth stimulatory effect, but also had an
invasiveness stimulatory effect on the progestin-resistant Ishikawa
cells. It has been reported that some patients who did not respond
to progestin treatment presented either invasion of the myometrium
and isthmus, lymph node invasion or pelvic metastases, at surgery
(1,5). The question of whether these
metastases existed before progestin treatment, whether the tumor
naturally progressed or whether progestin’s stimulatory effects
were involved in the process, requires further study.
Progesterone responses are rarely complete in
endometrial cancer patients with heterogeneous cancer cells
(11). Mechanisms must exist de
novo, or evolve during treatment, that allow a proportion of
cancer cells to escape progesterone inhibition and ultimately
support resistant growth. The progesterone-resistant cells selected
from the Ishikawa cell line by stepwise selection may be a
combination of primary and acquired drug resistance.
MPA could inhibit Ishikawa cell proliferation and
invasiveness, which suggests that using MPA as the conservative
treatment method in early-stage endometrial carcinoma is feasible
and effective. MPA has been used extensively in the treatment of
young patients with endometrial carcinoma as an endocrine therapy
with a response rate of approximately 57–75%. Moreover,
conservative treatment followed by IVF enables such patients to
achieve a successful pregnancy (1–5).
Unfortunately, progesterone-resistant cells could arise after
long-term treatment with MPA. When progesterone-resistant cell
lines arise, MPA treatment could stimulate proliferation and
metasis in these progestin-resistant cancer cells. In the period of
MPA treatment, we should be cautious of the phenomenon of
progesterone resistance.
The presence of the estrogen receptor (ER) and
progesterone receptor (PR) are well-known as prerequisites for
progestin action (12,13). To date, two ER and PR isoforms have
been identified: ERα, ERβ, PRA and PRB. ERα and ERβ share a high
degree of amino acid homology; however, there are significant
differences in regions of these receptors that would be expected to
influence transcriptional activity. ERα and ERβ form heterodimers
within target cells; ERβ modulates ERα transcriptional activity;
and high levels of ERα significantly inhibit the growth of tumors
and downregulate vascular endothelial growth factor expression in
tumors xenografted from the Ishikawa cells (14–16).
PRB is a 114-KDa protein, whereas PRA is a 94-KDa protein that
lacks 164 amino acids from the N-terminus. They are products of a
single gene and are translated from individual mRNA species under
the control of distinct promoters and have different functions. The
magnitude of transcriptional activation of PRB can be significantly
greater than that of PRA. It has been reported that
progesterone-inhibited cell growth and invasiveness may occur
mainly through PRB. Moreover, a drastic decrease of PRB reflects
poor prognosis in endometrial cancer patients (17–19).
The relative expression level of the ER and PR isoforms may be a
key determinant of cellular responses to endocrine treatment.
Compared with Ishikawa cells, progestin-resistant Ishikawa cells
have decreased the positive expression rate of ERα and PRB, and
increased the positive expression rate of ERβ. PRA remained
relatively constant over long-term treatment of MPA. Downregulation
of ERα, PRB and upregulation of ERβ may be involved in the
progestin resistance of endometrial carcinoma. Therefore, it is
important to examine ER and PR subsets when evaluating progesterone
effects. Abundant PRB, ERα expression in carcinoma cells may be a
necessary prerequisite for successful MPA treatment. Progesterone
resistance is a complex process; the change in ER and PR did not
inhibit the stimulatory effects of MPA on the progestin-resistant
cells. A compensatory pathway may therefore exist to support
proliferation over long-term MPA treatment (19–25).
This requires further study.
From the evidence, it might be concluded that
prolonged treatment with MPA in Ishikawa cells could give rise to a
resistant effect to MPA. When the resistance subtype is acquired,
treatment with MPA is capable of enhancing cancer cell
proliferation, invasiveness and metastasis. The imbalance of ER
subtype and PR subtype might contribute to the mechanisms involved
in the progesterone resistance. Determination of the subtype of ER
and PR may provide important additional information on the hormone
sensitivity of endometrial carcinoma.
Acknowledgements
The authors acknowledge the financial
support of The Foundation of Technological Innovation of Henan
Health (Zhengzhou, China).
References
|
1
|
Kim JJ and Chapman-Davis E: Role of
progesterone in endometrial cancer. Semin Reprod Med. 28:81–90.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jadoul P and Donnez J: Conservative
treatment may be beneficial for young women with atypical
endometrial hyperplasia or endometrial adenocarcinoma. Fertil
Steril. 80:1315–1324. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cade TJ, Quinn MA, Rome RM and Neesham D:
Progestogen treatment options for early endometrial cancer. BJOG.
117:879–884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Niwa K, Tagami K, Lian Z, Onogi K, Mori H
and Tamaya T: Outcome of fertility-preserving treatment in young
women with endometrial carcinomas. BJOG. 112:317–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laurelli G, Di Vagno G, Scaffa C, Losito
S, Del Giudice M and Greggi S: Conservative treatment of early
endometrial cancer: preliminary results of a pilot study. Gynecol
Oncol. 120:43–46. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murphy LC, Dotzlaw H, Wong MS, Miller T
and Murphy LJ: Mechanisms involved in the evolution of progestin
resistance in human breast cancer cells. Cancer Res. 51:2051–2057.
1991.PubMed/NCBI
|
|
7
|
Coldham NG, Patel K and Braunsberg H:
Hormone and cytotoxic drug responsiveness of cultured human breast
cancer cells resistant to specific hormones. Int J Cancer.
45:712–718. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herman ME and Katzenellembogen BS:
Response-specific antiestrogen resistance in a newly characterized
MCF-7 human breast cancer cell line resulting from long-term
exposure to trans-hydroxytamoxifen. J Steroid Biochem Mol Biol.
59:121–134. 1996. View Article : Google Scholar
|
|
9
|
Nishida M, Kasahara K, Kaneko M, Iwasaki H
and Hayashi K: Establishment of a new human endometrial
adenocarcinoma cell line, Ishikawa cells, containing estrogen and
progesterone receptors. Nippon Sanka Fujinka Gakkai Zasshi.
37:1103–1111. 1985.PubMed/NCBI
|
|
10
|
Ito K, Utsunomiya H, Yaegashi N and Sasano
H: Biological roles of estrogen and progesterone in human
endometrial carcinoma - new developments in potential endocrine
therapy for endometrial cancer. Endocr J. 54:667–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zaino RJ, Clarke CL, Mortel R and
Satyaswaroop PG and Satyaswaroop PG: Heterogeneity and
progesterone-receptor distribution in endometrial adenocarcinoma.
Cancer Res. 48:1889–1895. 1988.
|
|
12
|
Jongen V, Briët J, de Jong R, ten Hoor K,
Boezen M, van der Zee A and Nijman H: Expression of estrogen
receptor-alpha and -beta and progesterone receptor-A and -B in a
large cohort of patients with endometrioid endometrial cancer.
Gynecol Oncol. 112:537–542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stoian SC, Simionescu C, Mărgăritescu C,
Stepan A and Nurciu M: Endometrial carcinomas: correlation between
ER, PR, Ki67 status and histopathological prognostic parameters.
Rom J Morphol Embryol. 52:631–636. 2011.PubMed/NCBI
|
|
14
|
Hall JM and McDonnell DP: The estrogen
receptor beta-isoform (ERbeta) of the human estrogen receptor
modulates ERalpha transcriptional activity and is a key regulator
of the cellular response to estrogens and antiestrogens.
Endocrinology. 140:5566–5578. 1999.
|
|
15
|
Ali SH, O’Donnell AL, Mohamed S, Mousa S
and Dandona P: Overexpression of estrogen receptor-alpha in the
endometrial carcinoma cell line Ishikawa: inhibition of growth and
angiogenic factors. Gynecol Oncol. 95:637–645. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ali SH, O’Donnell AL, Balu D, Pohl MB,
Seyler MJ, Mohamed S, Mousa S and Dandona P: Estrogen
receptor-alpha in the inhibition of cancer growth and angiogenesis.
Cancer Res. 60:7094–7098. 2000.PubMed/NCBI
|
|
17
|
Miyamoto T, Watanabe J, Hata H, Jobo T,
Kawaguchi M, Hattori M, Saito M and Kuramoto H: Significance of
progesterone receptor-A and -B expressions in endometrial
adenocarcinoma. J Steroid Biochem Mol Biol. 2:110–118.
2004.PubMed/NCBI
|
|
18
|
Smid-Koopman E, Kuhne LC, Hanekamp EE,
Gielen SC, De Ruiter PE, Grootegoed JA, Helmerhorst TJ, Burger CW,
Brinkmann AO, Huikeshoven FJ and Blok LJ: Progesterone-induced
inhibition of growth and differential regulation of gene expression
in PRA- and/or PRB-expressing endometrial cancer cell lines. J Soc
Gynecol Invest. 12:285–292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai D, Wolf DM, Litman ES, White MJ and
Leslie KK: Progesterone inhibits human endometrial cancer cell
growth and invasiveness: down-regulation of cellular adhesion
molecules through progesterone B receptors. Cancer Res. 62:881–886.
2002.
|
|
20
|
Aghajanova L, Velarde MC and Giudice LC:
Altered gene expression profiling in endometrium: evidence for
progesterone resistance. Semin Reprod Med. 28:51–58. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arpino G, Weiss H, Lee AV, Schiff R, De
Placido S, Osborne CK and Elledge RM: Estrogen receptor-positive,
progesterone receptor-negative breast cancer: association with
growth factor receptor expression and tamoxifen resistance. J Natl
Cancer Inst. 97:1254–1261. 2005. View Article : Google Scholar
|
|
22
|
Wang S, Pudney J, Song J, Mor G, Schwartz
PE and Zheng W: Mechanisms involved in the evolution of progestin
resistance in human endometrial hyperplasia - precursor of
endometrial cancer. Gynecol Oncol. 88:108–117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang S, Thiel KW and Leslie KK:
Progesterone: the ultimate endometrial tumor suppressor. Trends
Endocrinol Metab. 22:145–152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Engelsen IB, Stefansson IM, Akslen LA and
Salvesen HB: GATA3 expression in estrogen receptor alpha-negative
endometrial carcinomas identifies aggressive tumors with high
proliferation and poor patient survival. Am J Obstet Gynecol.
199:543–547. 2008. View Article : Google Scholar
|
|
25
|
Rubel CA, Jeong JW, Tsai SY, Lydon JP and
Demayo FJ: Epithelial-stromal interaction and progesterone
receptors in the mouse uterus. Semin Reprod Med. 28:27–35. 2010.
View Article : Google Scholar : PubMed/NCBI
|