Introduction
Epigenetic modifications affect gene expression.
Tumors often show an aberrant epigenetic modification pattern,
including histone deacetylation and DNA hypermethylation, which
leads to the suppression of gene expression (1). Colorectal cancer (CRC) in particular
results not only from an accumulation of genetic changes, but also
epigenetic changes, which occur in the course of the transformation
of normal epithelium into adenocarcinomas.
Histone deacetylases (HDACs) catalyze the removal of
acetyl groups, thereby stimulating chromatin condensation, which
promotes transcriptional repression, including the shutdown of
tumor suppressor gene expression (2). HDACs are also able to target numerous
non-histone proteins, such as p53, even accounting for a majority
of HDAC substrates (3).
Overexpression of HDACs and epigenetic silencing has been
frequently observed in CRC (4,5).
Amongst others, these studies show that the perturbation of the
balance between acetylation and deacetylation plays an important
role in neoplastic transformation. As epigenetic changes are
reversible, HDACs constitute promising targets for pharmacological
inhibition in CRC (3).
By mechanisms not yet fully elucidated, HDAC
inhibitors are able to induce cell-cycle arrest, promote
differentiation and selectively stimulate apoptosis in transformed
cells via the extrinsic and/or intrinsic apoptotic pathway
(6). Furthermore, they
synergistically enhance the anticancer activity of chemotherapeutic
drugs, particularly those that are pro-apoptotic, by shifting the
balance between pro- and anti-apoptotic proteins.
Examples of HDAC inhibitors include short-chain
fatty acids, hydroxamic acids, benzamides and cyclic tetrapeptides
(2). The broad spectrum HDAC
inhibitor SAHA (also known as Zolinza® or Vorinostat)
was the first HDAC inhibitor to have successfully completed
clinical trials and is used for the treatment of cutaneous T-cell
lymphoma (7). Numerous HDAC
inhibitors are currently undergoing clinical trials (2). TSA is another hydroxamic acid and
broad spectrum HDAC inhibitor which targets class I, II and IV
HDACs in the same way as SAHA (3).
DNA hypermethylation is another frequent phenomenon
in cancer that silences many genes for cell cycle regulation,
receptors and apoptosis by DNA methylation of CpG islands in their
promoter region (8). A proportion
of proximal colon tumors (30–40%), and distal and rectal tumors
(3–12%), exhibit a CpG island methylator phenotype, where numerous
CpG islands are methylated and various tumor suppressor genes are
inactivated (9).
DNA methyltransferase (DNMT) inhibitors are able to
induce DNA demethylation and thus the reactivation of
epigenetically silenced genes. At present, there are two
FDA-approved drugs with DNMT inhibitory activity: 5-azacytidine and
decitabine, both of which are used in the treatment of
myelodysplastic syndrome and myeloid leukemias (3). Global reduction of DNA methylation has
been demonstrated to have anticancer effects in intestinal
tumorigenesis (10).
Histone deacetylation has been revealed to act
synergistically with DNA methylation in the epigenetic silencing of
cancer genes (11). Therefore
combinations of DNMT and HDAC inhibitors may have potential for
further clinical trials (10).
Natural substances have been found to act on
epigenetic signaling; green tea polyphenols demonstrated similar
effects to TSA in prostate cancer cells by inducing cell cycle
arrest and apoptosis (12). These
polyphenols were able to inhibit HDACs and induce their proteasomal
degradation. Furthermore, the tea polyphenol
(-)-epigallocatechin-3-gallate was also able to inhibit DNMT
activity and reactivate methylation-silenced genes in CRC cells
(13). In addition, apple
polyphenols reduced DNA methylation by inhibition of DNMT in CRC
cells (14). Moreover, soy
isoflavones reversed DNA hypermethylation and reactivated silenced
genes in esophageal squamous cell carcinoma cells (15), and their anticancer activity was
enhanced when used in combination with HDAC inhibitors (15). The polyphenol curcumin has also been
shown to inhibit DNMT activity (16) and to act synergistically with TSA in
inducing cell death (17).
The flavonolignan silibinin, which is the main
pharmacologically active component of the milk thistle plant
(Silybum marianum), has been shown to increase acetylation
of histones in hepatic cancer in vitro and in vivo.
Silibinin increased acetylation of histone H3 and H4 in
vitro in HuH7 cells (18), and
in vivo in HuH7 xenografts in nude mice (19). In non-small cell lung cancer,
silibinin inhibited HDAC activity and decreased HDAC levels
(20).
However, no studies have yet to fully describe the
effect of silibinin on DNMT activity. Previously, we demonstrated
that silibinin exerted anti-proliferative and pro-apoptotic effects
in the primary adenocarcinoma SW480 cells and in their metastatic
derivatives (SW620 cells) (21). In
the present study, we aimed to investigate whether silibinin
modified HDAC and DNMT activity in this model of CRC
progression.
Material and methods
Cell culture and treatment
SW480 and SW620 cells were purchased from the
European Collection of Animal Cell Culture (ECACC, Salisbury, UK).
The cells were cultured in 75 cm2 Falcon flasks in
Dulbecco’s modified Eagle’s medium (DMEM) containing 25 mM glucose
and supplemented with 10% heat-inactivated (56°C) horse serum, 100
U/ml penicillin, 100 μg/ml streptomycin and 1% non-essential
amino acids (Invitrogen Life Technologies, Lyon, France). Cells
were maintained at 37°C in a humidified atmosphere with 5%
CO2, and subcultured following trypsinization (0.5%
trypsin/2.6 mM ethylendiamine tetraacetic acid). For all
experiments, cells were seeded at 1×106 cells in culture
dishes (10 cm internal diameter). The culture medium used was DMEM
supplemented with 3% heat-inactivated horse serum and 100 U/ml
penicillin, 100 μg/ml streptomycin and 1% non-essential
amino acids. Additionally, 5 μg/ml transferrin, 5 ng/ml
selenium and 10 μg/ml insulin were added to compensate for
the lower serum concentration. The culture medium was replaced
every 48 h. Cells were exposed to silibinin (Sigma-Aldrich Chemie
GmbH, Steinheim, Germany) 24 h after seeding. Silibinin was
dissolved in dimethylsulf-oxide (DMSO; Sigma-Aldrich Chemie GmbH)
and used at a final concentration of 300 μM. The final
concentration of DMSO in the culture medium did not exceed 0.1%.
The HDAC inhibitors, TSA (0.1 μM) or SAHA (1 μM)
(Sigma-Aldrich Chemie GmbH), were added 1 h prior to silibinin
treatment.
Histone deacet ylase (HDAC) and DNA
methyltransferase (DNMT) activity
To determine the activity/inhibition of the
HDACs/DNMTs in the nuclear samples, the colorimetric EpiQuik™ HDAC
Activity/Inhibition Assay kit (Epigentek-Euromedex, Strasbourg,
France) and the colorimetric EpiQuik™ DNMT Activity/Inhibition
Assay Ultra kit (Epigentek-Euromedex) were used. Cells were
harvested by scraping, and nuclear extracts of the cells were
prepared using the EpiQuik™ Nuclear Extraction kit
(Epigentek-Euromedex). The protein content of the nuclear extracts
was determined by the Lowry assay, and any nuclear extracts not
immediately used were stored at −80°C.
To measure the HDAC activity, the same quantity of
nuclear extract was incubated with acetylated histone substrate in
a 96-well plate, and the quantity of remaining un-deacetylated
histone that is inversely proportional to HDAC enzyme activity in
the nuclear sample was colorimetrically quantified through an
ELISA-like reaction at a wavelength of 450 nm. The activity of HDAC
enzymes was inversely proportional to the OD.
To measure the DNMT activity, nuclear extracts were
incubated on a microplate stably coated with a universal DNMT
substrate. DNMT enzymes from the nuclear sample methylate the DNA
substrate during incubation, and then the quantity of methylated
DNA that is proportional to enzyme activity was colorimetrically
quantified through an ELISA-like reaction at a wavelength of 450
nm. The activity of DNMT enzymes was proportional to the OD.
Flow cytometric analysis of the sub-G0/G1
cell population
The sub-G0/G1 cell population (hypodiploid cells:
dying and dead cells) was analyzed by labeling cells with propidium
iodide. Cells were harvested by trypsinization after 24, 48 and 72
h of treatment and washed with phosphate-buffered saline (PBS; 0.1
M; pH 7.4). Cells were fixed in 70% ethanol at −20°C for ≥30 min,
washed twice with PBS and re-suspended in 200 μl PBS
containing 0.25 mg/ml RNase A and 0.1 mg/ml propidium iodide
(Sigma-Aldrich Chemie GmbH). Following incubation in the dark at
37°C for 30 min, the fluorescence of 10,000 cells/sample was
analyzed by flow cytometry, and histograms were analyzed using the
CellQuest software (FACScan, BD Biosciences, Belgium).
Statistical analysis
All experiments were performed ≥3 times. Data are
presented as mean ± standard error. Statistical differences between
the control and treated groups were evaluated using the Student’s
t-test or the Student-Newman-Keuls multiple comparison test.
P<0.05 was considered to indicate a statistically significant
difference between groups.
Results
Effects of silibinin on HDAC and DNMT
activity
To establish whether silibinin induced epigenetic
modifications in SW480 and SW620 cells, we determined HDAC and DNMT
activity in nuclear extracts of silibinin-treated cells with the
aid of the colorimetric EpiQuik HDAC Activity/Inhibition Assay kit
and the EpiQuik DNMT Activity/Inhibition Assay Ultra kit.
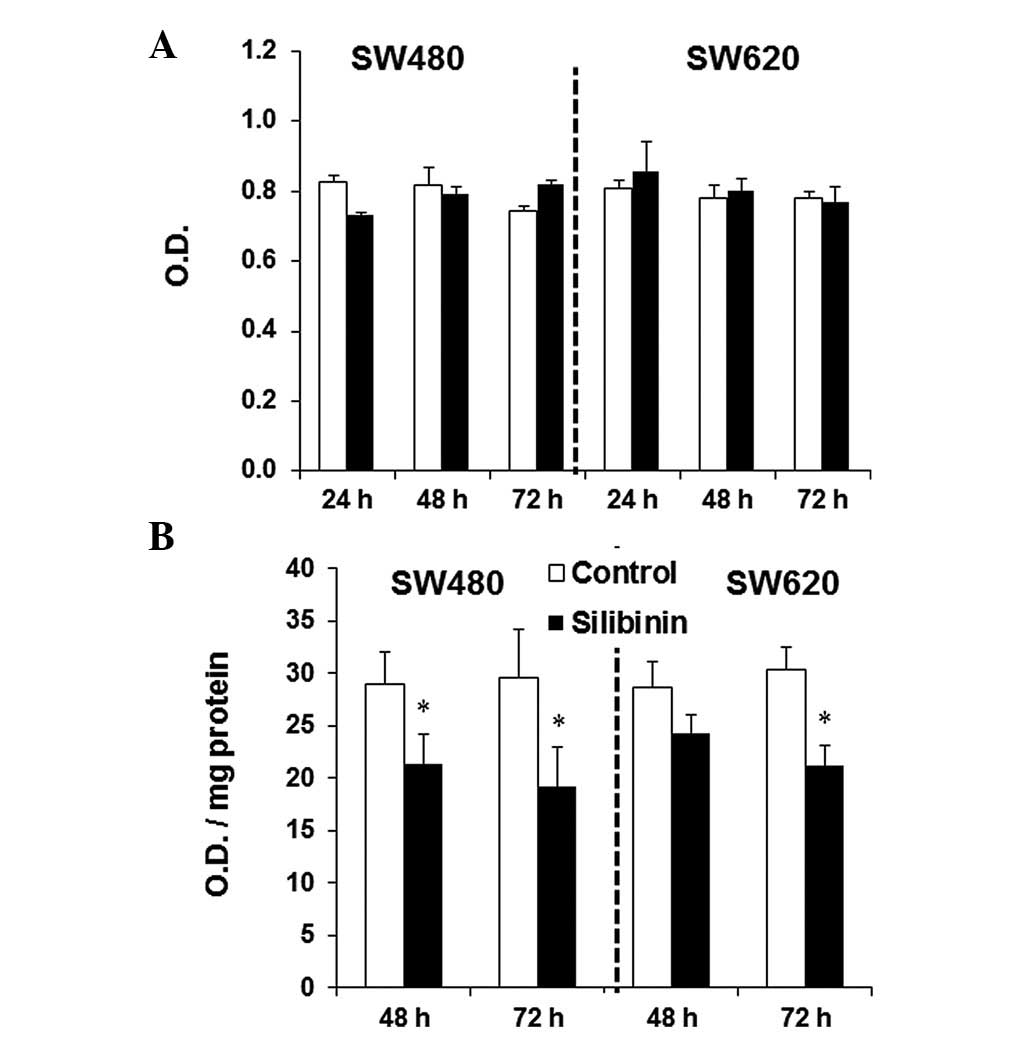
HDAC activity in SW480 and SW620 cells was not
changed by silibinin treatment (Fig.
1A). In contrast, a reduction in DNMT activity was observed in
silibinin-treated SW480 and SW620 cells after 48 h of treatment
with silibinin (Fig. 1B). However,
this reduction only became significant following 72 h of treatment
in SW620 cells.
Silibinin and HDAC inhibitors induce
synergistic cell death
Because synergy between DNA demethylation and HDAC
inhibition has been demonstrated in the re-expression of genes
silenced in cancer (11), we
investigated the synergistic effect of silibinin, which inhibits
DNMT as shown, in combination with HDAC inhibitors on cell
death.
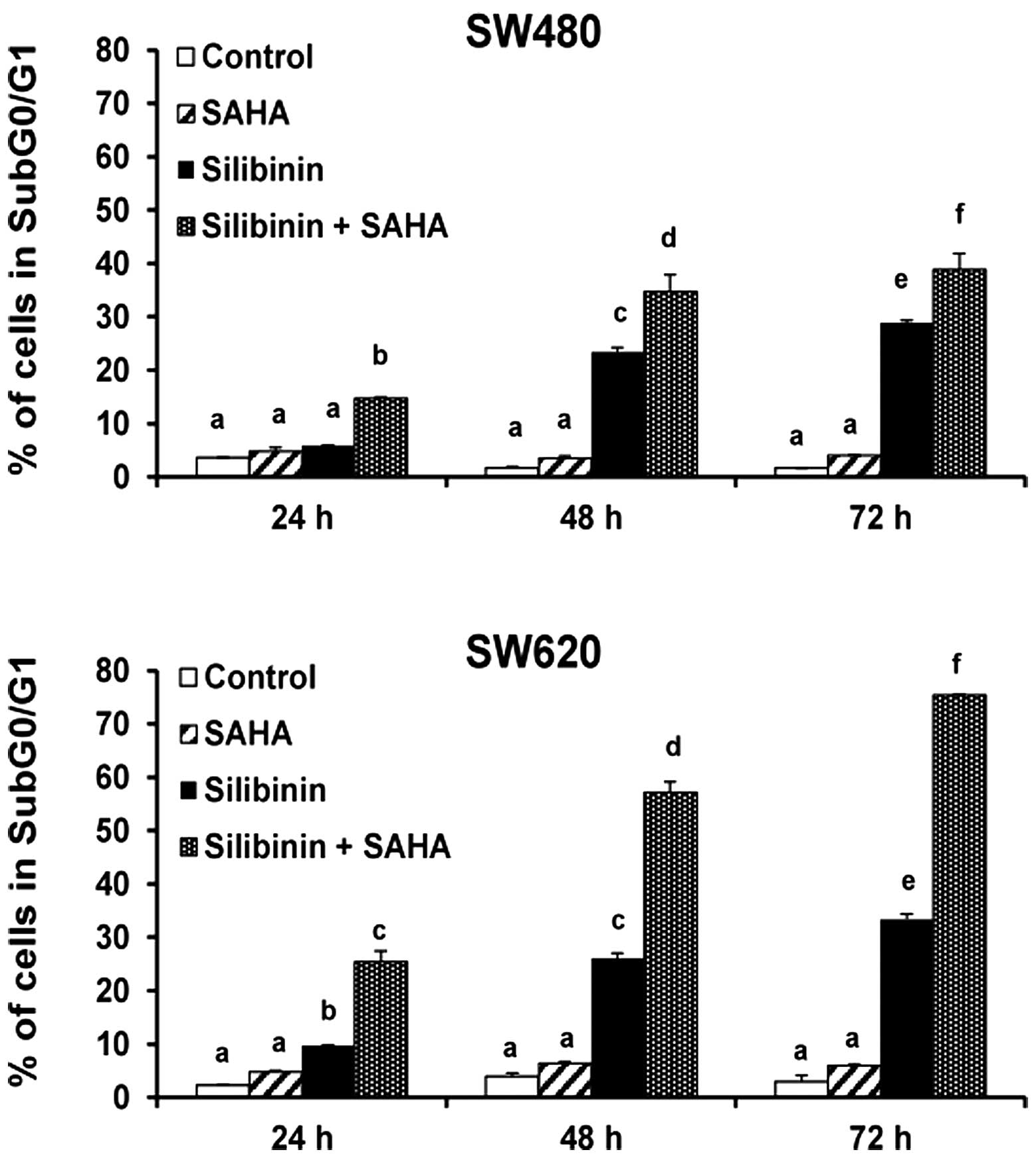
The clinically used HDAC inhibitor SAHA
significantly enhanced silibinin-induced cell death (Fig. 2) but demonstrated no cell toxicity
on its own. However, this effect was notably stronger in SW620
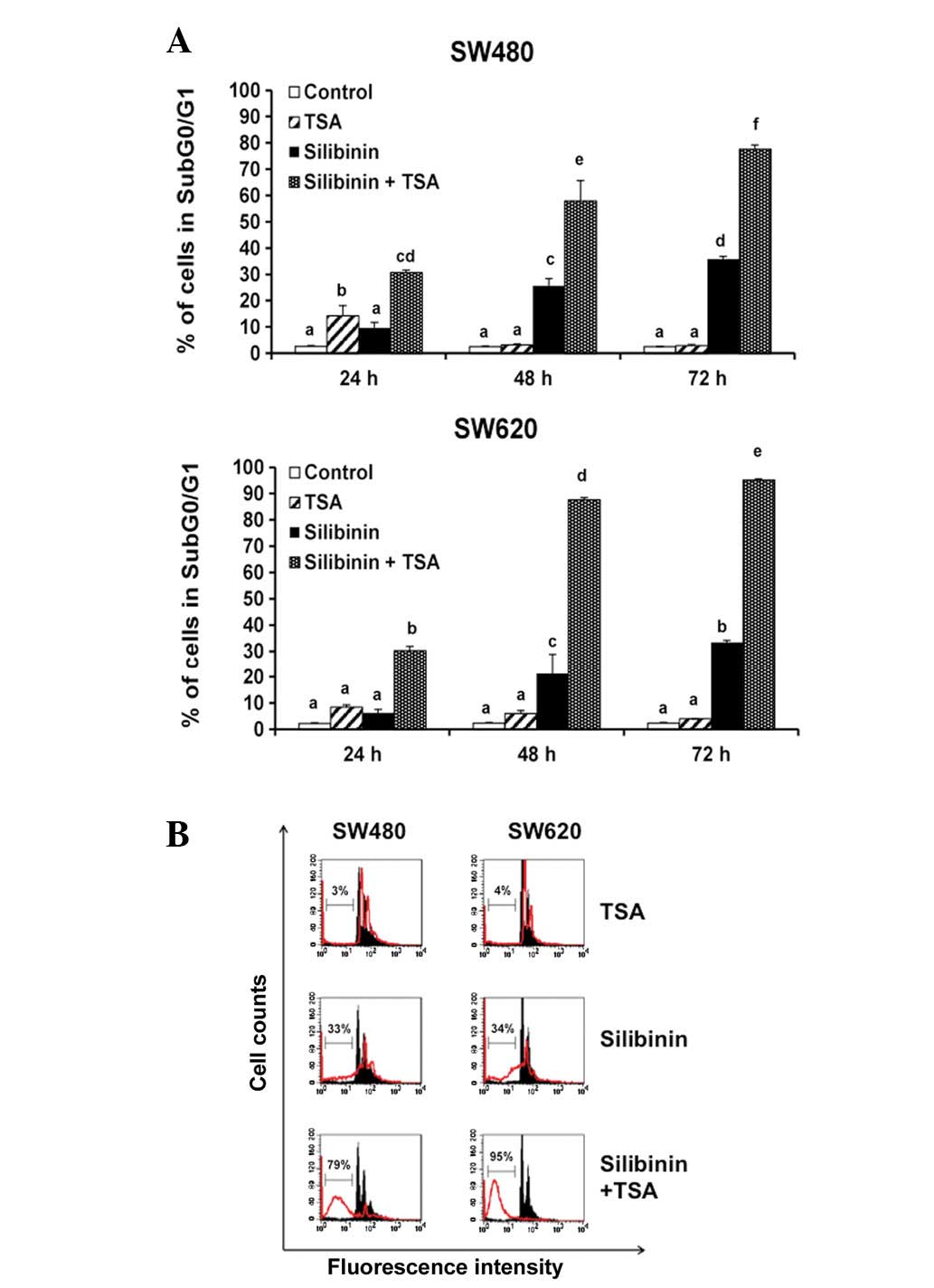
cells. To verify the interaction between silibinin and HDAC
inhibitors, we used the broad spectrum HDAC inhibitor TSA and
observed a clear synergistic effect with silibinin on cell death
induction in both cell lines (Fig.
3). Cell death induction was more notable with TSA than with
SAHA, and we found that the metastatic SW620 cells were more
sensitive to the combined treatment than the adenocarcinoma SW480
cells.
Discussion
Previously, we demonstrated that the polyphenol
silibinin inhibited cell growth and induced apoptosis in SW480 and
SW620 cells (21). In an
azoxymethane-induced rat model, silibinin was able to prevent the
formation of preneoplastic lesions, thus appearing to be a
promising chemopreventive agent in CRC (22). Polyphenols have been shown to modify
histone deacetylation and DNA hypermethylation, accompanying their
chemopreventive effect (12–14,16,20).
In the present study, we investigated the epigenetic
effects of silibinin. We found that silibinin did not change HDAC
activity in SW480 and SW620 cells. These data contrasted with
previous observations by Lah et al and Cui et al in
hepatocarcinoma cells and xenografts (18,19),
and by Mateen et al in non-small cell lung cancer (20), where silibinin was able to inhibit
the activity of HDACs. However, silibinin reduced DNMT activity in
both cell lines following 72 h of treatment. Inhibition of DNMT
activity was already significant at 48 h in SW480 cells.
As other polyphenols and DNMT inhibitors have been
demonstrated to act synergistically with HDAC inhibitors in cell
death induction, we tested the effect of a combination of silibinin
with two broad-spectrum HDAC inhibitors, SAHA and TSA, on the two
cell lines. Both combinations synergistically induced cell death.
These results were in agreement with those of other studies
demonstrating that silibinin synergistically augmented the
cytotoxic effects of SAHA and TSA in non-small cell lung cancer
cells (20). Notably, in our study,
SW480 and SW620 cells were both resistant to treatment by the HDAC
inhibitors, which alone exhibited no cytotoxic effects.
However, the synergistic effect of silibinin and
HDAC inhibitors could not be entirely attributed to
silibinin-induced DNMT inhibition, as the increase in cell death
occurred prior to significant DNMT inhibition by silibinin in SW620
cells. Furthermore, the synergy in cell death induction was
stronger in SW620 cells (up to 95% cell death compared with 80% in
SW480 cells), whereas DNMT inhibition was weaker in SW620 cells
than in SW480 cells.
Silibinin and HDAC inhibitors possess pleiotropic
anticancer activities, which may explain their synergistic effects;
the ability of HDAC inhibitors to change the balance between pro-
and anti-apoptotic factors (6) may
contribute to the enhancement of the apoptosis-inducing properties
of silibinin.
In conclusion, silibinin inhibited DNMT but not HDAC
activity in colorectal SW480 and metastatic SW620 cells, and
exerted synergistic effects with HDAC inhibitors on cancer cell
death. Further investigations are required to determine the
mechanisms involved in this process. However, our data suggest that
treatments combining silibinin and HDAC inhibitors may represent a
promising approach, given the non-toxic nature of silibinin and the
fact that HDAC inhibitors selectively target cancer cells. Combined
treatment of silibinin with different epigenetic agents, including
HDAC inhibitors, in current clinical trials may thus contribute to
the development of novel combination therapies.
Acknowledgements
Henriette Kauntz was supported by a
fellowship provided by the Conseil Régional d’Alsace, France.
References
|
1
|
Lu Q, Qiu X, Hu N, Wen H, Su Y and
Richardson BC: Epigenetics, disease, and therapeutic interventions.
Ageing Res Rev. 5:449–467. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song SH, Han SW and Bang YJ:
Epigenetic-based therapies in cancer: progress to date. Drugs.
71:2391–2403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carew JS, Giles FJ and Nawrocki ST:
Histone deacetylase inhibitors: mechanisms of cell death and
promise in combination cancer therapy. Cancer Lett. 269:7–17. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mariadason JM: HDACs and HDAC inhibitors
in colon cancer. Epigenetics. 3:28–37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ashktorab H, Belgrave K, Hosseinkhah F,
Brim H, Nouraie M, Takkikto M, Hewitt S, Lee EL, Dashwood RH and
Smoot D: Global histone H4 acetylation and HDAC2 expression in
colon adenoma and carcinoma. Dig Dis Sci. 54:2109–2117. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Emanuele S, Lauricella M and Tesoriere G:
Histone deacetylase inhibitors: apoptotic effects and clinical
implications (Review). Int J Oncol. 33:637–646. 2008.PubMed/NCBI
|
|
7
|
Wagner JM, Hackanson B, Lübbert M and Jung
M: Histone deacetylase (HDAC) inhibitors in recent clinical trials
for cancer therapy. Clin Epigenetics. 1:117–136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
9
|
Migliore L, Migheli F, Spisni R and
Coppedè F: Genetics, cytogenetics, and epigenetics of colorectal
cancer. J Biomed Biotechnol. 2011:7923622011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rius M and Lyko F: Epigenetic cancer
therapy: rationales, targets and drugs. Oncogene. 31:4257–4265.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cameron EE, Bachman KE, Myöhänen S, Herman
JG and Baylin SB: Synergy of demethylation and histone deacetylase
inhibition in the re-expression of genes silenced in cancer. Nat
Genet. 21:103–107. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thakur VS, Gupta K and Gupta S: Green tea
polyphenols causes cell cycle arrest and apoptosis in prostate
cancer cells by suppressing class I histone deacetylases.
Carcinogenesis. 33:377–384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H,
Welsh W and Yang CS: Tea polyphenol (-)-epigallocatechin-3-gallate
inhibits DNA methyltransferase and reactivates methylation-silenced
genes in cancer cell lines. Cancer Res. 63:7563–7570.
2003.PubMed/NCBI
|
|
14
|
Fini L, Selgrad M, Fogliano V, Graziani G,
Romano M, Hotchkiss E, Daoud YA, De Vol EB, Boland CR and
Ricciardiello L: Annurca apple polyphenols have potent
demethylating activity and can reactivate silenced tumor suppressor
genes in colorectal cancer cells. J Nutr. 137:2622–2628. 2007.
|
|
15
|
Fang MZ, Chen D, Sun Y, Jin Z, Christman
JK and Yang CS: Reversal of hypermethylation and reactivation of
p16INK4a, RARbeta, and MGMT genes by genistein and other
isoflavones from soy. Clin Cancer Res. 11:7033–7041. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shu L, Khor TO, Lee JH, Boyanapalli SSS,
Huang Y, Wu TY, Saw CL, Cheung KL and Kong AN: Epigenetic CpG
demethylation of the promoter and reactivation of the expression of
Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 13:606–614.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan G, Graham K and Lanza-Jacoby S:
Curcumin enhances the anticancer effects of trichostatin a in
breast cancer cells. Mol Carcinog. Jan 30–2012.(Epub ahead of
print).
|
|
18
|
Lah JJ, Cui W and Hu KQ: Effects and
mechanisms of silibinin on human hepatoma cell lines. World J
Gastroenterol. 13:5299–5305. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cui W, Gu F and Hu KQ: Effects and
mechanisms of silibinin on human hepatocellular carcinoma
xenografts in nude mice. World J Gastroenterol. 15:1943–1950. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mateen S, Raina K, Jain A, Agarwal C, Chan
D and Agarwal R: Epigenetic modifications and p21-cyclin B1 nexus
in anticancer effect of histone deacetylase inhibitors in
combination with silibinin on non-small cell lung cancer cells.
Epigenetics. 7:1161–1172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kauntz H, Bousserouel S, Gossé F and Raul
F: Silibinin triggers apoptotic signaling pathways and autophagic
survival response in human colon adenocarcinoma cells and their
derived meta-static cells. Apoptosis. 16:1042–1053. 2011.
View Article : Google Scholar
|
|
22
|
Kauntz H, Bousserouel S, Gossé F,
Marescaux J and Raul F: Silibinin, a natural flavonoid, modulates
the early expression of chemoprevention biomarkers in a preclinical
model of colon carcinogenesis. Int J Oncol. 41:849–854.
2012.PubMed/NCBI
|