Introduction
Hereditary breast and ovarian cancer (HBOC) is an
autosomal dominant syndrome with incomplete penetrance. The two
most commonly mutated genes in HBOC are BRCA1 and
BRCA2, which are essential components of the double-strand
break repair system (1). Almost
3,500 cancer-associated mutations, scattered throughout the two
genes, have so far been reported in the Breast Cancer Information
Core (BIC) database (http://research.nhgri.nih.gov/bic/). The present study
reports a new germline nucleotide 3020insCT/c.2901insCT mutation
detected in the BRCA1 gene. In general, germline mutations
in known breast cancer risk genes account for ~20% of breast
cancers associated with a family history. It is therefore crucial
to identify these individuals to offer appropriate cancer
management and understand the contribution of BRCA1 and
BRCA2 mutation-associated risks.
Case report
A 55-year-old non-Ashkenazi Spanish female diagnosed
with breast cancer (at 51 years old) and ovarian cancer (at 55
years old) and treated at the University Clinic of Navarra (CUN;
Pamplona, Navarra, Spain), was transferred to the genetic
counseling unit. The clinical history of the patient lead us to
consider the possibility of HBOC syndrome. Following verbal and
written informed consent, genomic DNA was extracted from a
peripheral blood sample and the BRCA1 and BRCA2 genes
were sequenced on an automated analyzer (ABI PRISM®
3130XL; Applied Biosystems, Foster City, CA, USA). The results were
compared to the consensus wild-type sequences (Genbank NM_007294.2
for BRCA1 and Genbank NM_000059.1 for BRCA2). A
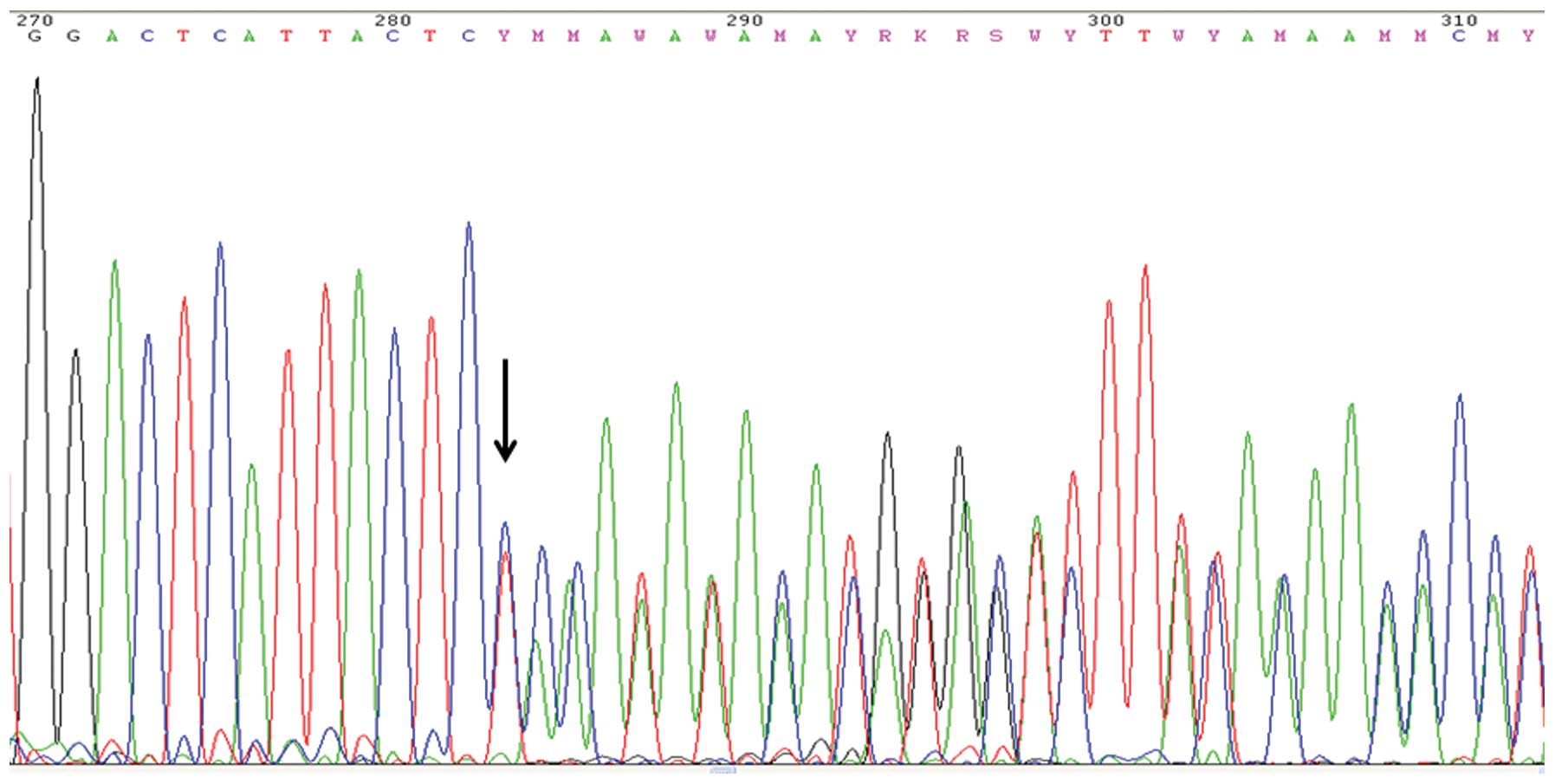
3020insCT/c.2901insCT frameshift mutation was identified in exon 11
of BRCA1 (Fig. 1). The
insertion was confirmed by repeated analyses including
reverse-primer sequencing. A BRCA1-Multiplex
Ligation-dependent Probe Amplification (MLPA) analysis was
performed, in order to investigate whether the mutation was able to
lead to an exon rearrangement. The results indicated that none of
the BRCA1 alleles showed deletion and/or duplication
(results not shown).
Genetic analysis was recommended to the only other
individual at risk in patient’s family, namely the twin sister, and
the analysis showed that she did not carry the mutation.
Discussion
A 3020insCT/c.2901insCT frameshift mutation in exon
11 of the BRCA1, which has yet to be reported in the BIC
database, was detected in a 55-year-old non-Ashkenazi Spanish
female diagnosed with breast and ovarian cancer. Since other family
members were not available for genetic analysis, the segregation of
the mutation could not be established. From the literature
available, it may be deduced that the mutation leads to the
deletion of the coiled-coil domain and BRCA1 C terminus (BRCT)
domains of the BRCA1 protein. The coiled-coil region is critical
for transcriptional activation through its interaction with the
basic leucine zipper (bZIP) domain of the JunB protein. In
vitro and in vivo experiments suggest that this
BRCA1-JunB interaction is particularly important for the
suppression of ovarian cancer (2).
The lack of the coiled-coil domain in the present patient may have
been closely correlated with the development of the ovarian cancer.
However, BRCA1 has a pivotal function within the BRCA1-associated
genome surveillance complex through the coordination of the actions
of damage-sensing and executive repair proteins. Solyom et
al(3) showed that the Abraxas
protein serves as a central organizer of a large BRCA1 holoenzyme
complex. Abraxas directly binds, via its phosphorylated C terminus,
to the BRCA1 BRCT motifs, linking BRCA1 to a core protein complex
dedicated to ubiquitin chain recognition and hydrolysis at DNA
double-strand breaks (3,4). Moreover, BRCT domains in BRCA1 are
able to bind DNA strand breaks and ends in vitro, which is
enhanced by the formation of the BRCA1-BARD1 heterodimer (5). The structural studies of Kobayashi
et al showed that the BRCT domain partially inserts into the
major groove and makes extensive contacts with the DNA backbone
(6), suggesting the possibility
that proteins with BRCT domains may act as DNA sensors and
transducers of DNA damage response signaling. The mutation
identified in the present study would markedly compromise these
functions, with profound biological consequences. The premature
stop codon at amino acid 1000 leads to a truncated protein that has
70% of its normal length. The advantage of having mutant
BRCA1 human breast cancer cell lines is that the impact of
pathogenic human mutations may be evaluated in the context of a
human genetic background. A previous study of 41 human breast
cancer cell lines identified a BRCA1 mutant cell line,
SUM149PT, with a nucleotide deletion at position 2288 (7). The resulting truncated BRCA1 protein
lacked the C-terminal BRCT and coiled-coil domains similar to the
present patient. Nuclear BRCA1 protein expression was not
detectable in the cell line, therefore corroborating the tumor
suppressor function of BRCA1 and the pathogenicity of the
mutation.
The next step was to search for bibliographic
evidence of the present mutation in BRCA1-knockout animal
models. The homozygous loss of BRCA1 generally leads to
early embryonic lethality, although it is possible to extend the
viability though the removal of p53 function. Among the range of
models available, McCarthy et al designed truncated human
BRCA1f22-24/p53+/− mice (harboring the second
BRCT domain), that develop estrogen receptor-negative
(ER−) and progesterone receptor-negative
(PR−) tumors lacking HER2 protein overexpression and
gene amplification (8). This
phenotype is similar to 64–90% of human BRCA1-mutation
breast cancers, so called ‘triple negative’ breast cancers. The
immunophenotypic features of the present patient’s tumor indicated
a noticeably different pattern, being ER+,
PR+ and ErbB2-negative. It has been reported that 10–36%
of BRCA1 mutation-related invasive breast cancers are, in
fact, ER+. Furthermore, BRCA1 mutation carriers
who are older or post-menopausal at the time of the diagnosis of
breast cancer are more likely to have an ER+ breast
cancer (9,10). With regard to the origin of these
ER+BRCA1-related breast cancers, Lim et al
observed the expansion of a committed luminal progenitor
population, containing ER+ and ER− cells, in
preneoplastic tissues of BRCA1 mutation carriers and
proposed the luminal progenitor cells as the cell of origin for
BRCA1-associated cancers (11). In mouse models with the deletion of
BRCA1, the expression of ER in the resulting tumors appears
to depend on whether BRCA1 is deleted at an earlier or later
stage of cell differentiation (12–14).
These studies suggest that BRCA1-deficient ER+
tumors may derive from BRCA1 loss in an ER+
luminal progenitor cell.
Another key point is the therapeutic approach for
ER+BRCA1-associated breast cancers. Given the
availability of effective therapies that exploit defects in
homologous recombination, such as PARP-1 inhibitors and cisplatin,
it is increasingly important to determine whether these therapies
are likely to be effective in ER+BRCA1-mutant
cancers. A recent study by Kaplan et al indicated that
ER+BRCA1-related breast cancers are
indistinguishable from ER−BRCA1-related cancers
in their nuclear expression of PARP-1, suggesting that
ER+BRCA1-related breast cancers may respond well
to drugs that exploit BRCA1 deficiency (15). ER+BRCA1-related
breast cancers appear to be a unique group and efforts should be
made to identify the individuals for whom estrogen-modifying agents
are likely to be particularly effective.
Acknowledgements
The authors are grateful to all patients and their
families.
References
|
1
|
Narod SA and Foulkes WD: BRCA1 and BRCA2:
1994 and beyond. Nat Rev Cancer. 4:665–676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hu YF and Li R: JunB potentiates function
of BRCA1 activation domain 1 (AD1) through a coiled-coil-mediated
interaction. Genes Dev. 16:1509–1517. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Solyom S, Aressy B, Pylkäs K, et al:
Breast cancer-associated abraxas mutation disrupts nuclear
localization and DNA damage response functions. Sci Transl Med.
4:122–123. 2012.PubMed/NCBI
|
|
4
|
Wang B, Matsuoka S, Ballif BA, et al:
Abraxas and RAP80 form a BRCA1 protein complex required for the DNA
damage response. Science. 316:1194–1198. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Simons AM, Horwitz AA, Starita LM, et al:
BRCA1 DNA-binding activity is stimulated by BARD1. Cancer Res.
66:2012–2018. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kobayashi M, Ab E, Bonvin AM and Siegal G:
Structure of the DNA-bound BRCA1 C-terminal region from human
replication factor C p140 and model of the protein-DNA complex. J
Biol Chem. 285:10087–10097. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Elstrodt F, Hollestelle A, Nagel JH, et
al: BRCA1 mutation analysis of 41 human breast cancer cell lines
reveals three new deleterious mutants. Cancer Res. 66:41–45. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McCarthy A, Savage K, Gabriel A, Naceur C,
Reis-Filho JS and Ashworth A: A mouse model of basal-like breast
carcinoma with metaplastic elements. J Pathol. 211:389–398. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Foulkes WD, Metcalfe K, Sun P, et al:
Estrogen receptor status in BRCA1- and BRCA2-related breast cancer:
The influence of age, grade, and histological type. Clin Cancer
Res. 10:2029–2034. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tung N, Wang Y, Collins LC, et al:
Estrogen receptor positive breast cancers in BRCA1 mutation
carriers: Clinical risk factors and pathologic features. Breast
Cancer Res. 12:R122010. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lim E, Vaillant F, Wu D, et al: Aberrant
luminal progenitors as the candidate target population for basal
tumor development in BRCA1 mutation carriers. Nat Med. 15:907–913.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poole AJ, Li Y, Kim Y, Lin SC, Lee WH and
Lee EY: Prevention of Brca1-mediated mammary tumorigenesis in mice
by a progesterone antagonist. Science. 314:1467–1470. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu S, Ginestier C, Charafe-Jauffret E, et
al: BRCA1 regulates human mammary stem/progenitor cell fate. Proc
Natl Acad Sci USA. 105:1680–1685. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ginestier C, Liu S and Wicha MS: Getting
to the root of BRCA1-deficient breast cancer. Cell Stem Cell.
5:229–230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kaplan JS, Schnitt SJ, Collins LC, et al:
Pathologic features and immunophenotype of estrogen
receptor-positive breast cancers in BRCA1 mutation carriers. Am J
Surg Pathol. 36:1483–1488. 2012. View Article : Google Scholar : PubMed/NCBI
|