Introduction
Xeroderma pigmentosum (XP) is a rare autosomal
recessive disorder characterized by extreme sensitivity to
sunlight, resulting in sunburn, pigmentation and cancer of exposed
skin, particularly in the oral maxillofacial region (1). XP has eight subtypes based on the gene
that is affected, namely XP-A, XP-B, XP-C, XP-D, XP-E, XP-F, XP-G
and Xeroderma pigmentosum variant (XP-V) (2). XP-V is particularly common, with an
incidence rate of 21% (3). Clinical
features of XP-V (MIM 278750) include sensitivity to sunlight and
sunlight-induced lesions. Significant pigmentation, dryness and
roughness commonly occur in sun-exposed skin. Photophobia and
corneal opacification are often present in patients (2). The greatest danger for the survival of
XP patients is the recurrence of cancer (1). Pigmentation in exposed areas
accelerates the malignancy of melanocytes and keratinocytes and
eventually leads to tumors, such as multiple basal and invasive
squamous cell carcinomas and melanomas (3).
Since mutations in the POLH gene (MIM 603968)
were first reported in XP-V cell lines in 1999, a number of
mutations in the POLH gene have been identified in XP-V
patients from various populations (4–13). The
human POLH gene encodes polymerase η (pol η), a homolog of
the yeast Rad30 protein, which belongs to the polymerase family Y
and is key in translesion DNA synthesis (TLS) (14,15).
Although XP-V patients often suffer from a number of various types
of skin cancer and a single case usually has more than one category
of tumor (8), little is known
concerning factors affecting tumor proneness in XP-V besides the
POLH mutation. Previous studies have revealed that the
expression of numerous oncogenes are altered in XP skin tumors
(16), which has indicated that
XP-V tumor development may involve the unusual expression of
numerous genes as well (2).
However, to date, no previous studies have explored the expression
patterns of other genes besides POLH and POLI in XP-V
tumors (6). XP-V skin cancer
proneness is mainly due to genetic instability caused by defective
TLS (17), and polymerases, such as
pol ζ, κ and ι, are responsible for cyclobutane pyrimidine dimer
(CPD) TLS (18). An additional
polymerase, pol θ, has the same function as pol η, which is to
generate A/T mutations during somatic hypermutation of Ig genes
(19). As all these polymerases
have been confirmed to bypass DNA damage, with functions similar to
those of pol η, it may be deduced that the genes REV3L,
POLK, POLI and POLQ, encoding pol ζ, κ, ι and
θ, respectively, may also have altered expression and contribute to
mutagenesis in XP-V tumors.
The present study reports a novel POLH
mutation and its associated phenotypic features in an XP-V patient,
as well as the expression of genes encoding pol η, ζ, κ, ι and θ in
XP-V lip tumor cells, XP-V cell lines and HeLa cells in which
POLH has been knocked down. The observations indicate a
possible correlation between several TLS polymerases and genetic
instability in XP-V tumor proneness.
Materials and methods
Enrollment of subjects
The study protocol was approved by the institutional
review board at the School of Stomatology, Wuhan University (Wuhan,
China) and informed written consent was obtained from all
participants or their guardians. A 65-year-old male was diagnosed
with XP disease at the Hospital of Stomatology, Wuhan University.
XP-V tumor cells (epithelial cells) were obtained from the lip
tumor (squamous cell carcinoma) of the patient, and control
epithelial cells were obtained from the normal lip tissue of three
children undergoing cleft lip repair surgery at the Hospital of
Stomatology, Wuhan University. Blood samples (1–3 ml) were obtained
from the diagnosed patient and 96 normal individuals from the local
population.
Polymerase chain reaction (PCR) and DNA
sequencing
Genomic DNA was isolated by the standard SDS
proteinase K salt chloroform technique (20). Based on a candidate gene approach,
all exons, candidate introns and the majority of flanking sequences
(GenBank POLH accession, NG_009252) were amplified by PCR.
Primers used for PCR were made by Sangon Biotech Co., Ltd.
(Shanghai, China). The sequence of primers are listed in Table I and the distribution of primers for
candidate introns are shown in Fig.
2C. PCR products were analyzed by 2% agarose gel
electrophoresis in 0.5X TBE buffer (Bioprimacy Co., Ltd., Wuhan,
China) at 5 V/cm and were sequenced by an ABI 3730xl DNA analyzer
(Applied Biosystems, Carlsbad, CA, USA).
 | Figure 2PCR results analyzed by gel
electrophoresis and the distribution of primers for long exons and
candidate introns. (A) Results of PCR amplification of POLH
exons analyzed by agarose gel electrophoresis showed that the
patient had no PCR products between exons 5 and 9. The upper lanes
represent the normal controls while the lower lanes represent the
XP-V patient. Lanes 1, DNA maker; 2–11, PCR products of exon 1–10;
and 12–15, four PCR products of four pairs of primers for exon 11.
(B) PCR products of introns 4 and 9 analyzed by agarose gel
electrophoresis. The upper lanes represent normal controls and the
lower lanes represent the XP-V patient. Lanes 1, DNA maker; 2–5,
PCR products of primers 4-1 to 4-4 in intron 4; and 6–8, PCR
products between primers 9-1 and 9-3 in intron 9. No PCR products
were formed for primers 4-3, 4-4 and 9-1 in the patient’s sample.
(C) Distribution of primers in introns 4 and 9 and exon 11. Gray
blocks indicate coding exons and white blocks indicate UTR regions,
while gray lines indicate introns. Each thick arrow represents a
primer and the numbers between the arrows correspond with the
number of primers in Table I. The
sites of arrows on the DNA strand indicate the approximate sites of
the primer binding on the DNA sequence. PCR, polymerase chain
reaction; XP-V, xeroderma pigmentosum variant. |
 | Table IPCR primers of POLH genomic
DNA. |
Table I
PCR primers of POLH genomic
DNA.
| Exon or intron
no. | Size, bp | Forward primer,
5′→3′ | Reverse primer,
5′→3′ |
|---|
| 1 | 554 |
GCTGGAGGAGGAGCGTTAGC |
ACCTTGGCCCAGTCACTGCT |
| 2 | 518 |
TCCATGCTCCCATGCTCATGG |
TCCCCATCTCTACCCACCCC |
| 3 | 574 |
CTGAACTGCTTTGTTTTGGATTGAA |
TGGATGGAGAACATGGGAATTGG |
| 4 | 568 |
GTTTGCCTCCGTGATTCTTCCT |
TCCTTCCATGTGAGTCCTTGTG |
| 5 | 448 |
GCCTATGCTGAAGCTAAGCTGC |
TTTCAATGTCCCACTGTCCCT |
| 6 | 397 |
CCCACTGGGGATGTTGTGGG |
TAAGGACTGGAGCCAGGGGA |
| 7 | 398 |
TGTCCTGAACCTTTTGGAGAGCT |
TGGGTCACTATGGCCCATCAG |
| 8 | 343 |
GGCAGGGGTTTCGTCAGAGG |
CCAAAACCTACCCACTGACCCC |
| 9 | 458 |
ACCTGATGGCAAACAAGC |
CTGGGAGACAGAGTGAGACC |
| 10 | 471 |
CCAAACCATTGTCACCCTGG |
TTACCCTTTACCTCATTGAAGGACT |
| 11-1 | 749 |
GGTTCTCAAGACATAACATCAGCA |
AAACAGGGACACACCCTGGA |
| 11-2 | 543 |
CAGGGAAGTGGCCCAGCG |
TACCGGTACCAGGGAGCCAC |
| 11-3 | 741 |
GTCAAAGTACAGGAACTGAGCCCT |
GCCTGTGAAGAGATGGGACCG |
| 11-4 | 417 |
ACCCCCAGGTTGTTTCTGCC |
AGACTCCAAGGCCCACACAC |
| 4-1 | 1333 |
AGGGAAGCTTGTGACTTAAGGAAT |
CTGAGCATGATTGCTAGCTCTTAT |
| 4-2 | 1766 |
ATAAGAGCTAGCAATCATGCTCAG |
TTAACTGAACGGGACCACAGA |
| 4-3 | 910 |
TATTTCCTGTAGTCCATTTCATGGTA |
ATTGTTAAAAGAACATTCTGCAGTCA |
| 4-4 | 1708 |
GTTCCTCAGCACAATGGCTTGC |
CCCGCAGCTTAGCTTCAGCA |
| 9-1 | 1813 |
GTACAATGGTGGCTGTTGCA |
GCTACAATACGGCTGAACCTG |
| 9-2 | 1976 |
CAGGTTCAGCCGTATTGTAGC |
GCAAAGAAACGGGAAGTGCT |
| 9-3 | 1614 |
AGCACTTCCCGTTTCTTTGC |
CTGAGGCGTTTGTCTCCTTG |
Cell culture and cDNA extraction
All cells, including XP-V tumor cells, normal cells,
XP-V fibroblast cell lines, human skin fibroblast (HSF) cell lines
and HeLa cells, were cultured in DMEM supplemented with 20% FBS
(HyClone, Logan, UT, USA). HeLa and HSF cells were purchased from
the cell bank of the Chinese Academy Of Sciences (Beijing, China).
XP-V fibroblast cell lines (XP30RO and XP1SF) were purchased from
Coriell Institute (Camden, NJ, USA). The cells were incubated at
37°C in 5% CO2 and the total RNA kit (Omega Bio-Tek,
Norcross, GA, USA) was used to extract RNA from each sample. The
RNA was used to synthesize cDNA using the First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA).
Vector construction, transfection and
fluorescence detection for subcellular localization
The following primers were purchased from Sangon
Biotech Co., Ltd., and were used to amplify the POLH coding
cDNA (GenBank POLH accession, NM_006502) from XP-V tumor cells and
control cells: Forward (F): 5′-ATGCTCGAGCAATGGCTACTGGAC AGGATCG-3′
and reverse (R): 5′-ACGGAATTCCCTGAG GGCAGCACTAATGT-3′. The mutant
and wild-type POLH cDNA were separately inserted into
pEYFP-C1 vectors (Clontech Laboratories, Inc., Mountain View, CA,
USA). The vectors were separately transfected into HeLa cells using
Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA,
USA) to test whether the mutant pol η had altered subcellular
localization. Visualization of YFP-tagged pol η was performed under
a fluorescence microscope (DM4000D; Leica, Wetzlar, Germany) at 48
h following transfection. Cells were stained with Hoechst 33258 to
mark the nuclei (Sigma-Aldrich, St. Louis, MO USA).
Knockdown of POLH expression in HeLa
cells
Expression of DNA pol η in HeLa cells was knocked
down by transfection with polymerase-specific siRNA with the
following sequence: 5′-GCCCUUCUUUAAGCAGAAATT-3′. siRNA were
purchased from GenePharma Co., Ltd. (Shanghai, China).
Real-time PCR (qPCR)
POLH, POLI, POLK, POLQ
and REV3L genes encoding the DNA polymerases pol η, ι, κ, θ
and ζ, respectively, were examined. Specifically, the expression
levels of these genes were compared between XP-V tumor cells and
normal control cells. In addition, changes in the expression of
these genes upon UV irradiation were also assayed. GAPDH was
used as an internal control for the normalization of RNA levels.
HeLa cells, with and without POLH knockdown, were used to
verify the changes in expression. For UV irradiation, cells were
exposed to UV-C light (5 J/m2) and were continued to be
cultured normally for 24 h thereafter. The dose of irradiation was
selected according to its effect on cells, which has evident
toxicity but maintains an appropriate level of cell activity
(8,18). The mRNA of HeLa cells with
POLH knockdown were isolated 24 h following transfection
with siRNA. Cells that received UV irradiation were irradiated at
24 h following siRNA transfection, when the expression of
POLH had evidently been knocked down. Cell mRNA was isolated
at 24 h following UV irradiation. Each sample was tested in
triplicate. All data are expressed as the mean ± standard error.
qPCR was performed using the SYBR® Premix Ex Taq™ II
reagent kit (Takara Bio, Inc., Shiga, Japan). The ABI 7500
Real-time PCR System (Applied Biosystems) was used to analyze the
data by the ΔΔCt method as described previously (21). To identify the differences in gene
expression, samples of tumor and HeLa cells without POLH
knockdown were defined as the reference samples, and the mRNA
quantity of all tested genes in the reference sample was defined as
‘1.0’. Student’s t-test was used to compare the relative expression
levels between tumor and normal cells and between HeLa cells with
and without POLH knockdown. Statistical analyses were
performed by SPSS (SPSS Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference. The
following primers were used to analyze the mRNA levels of the five
genes studied: F: 5′-AGTTCGTGAGTCCCGTGGG-3′ and R: 5′-GCTTGGCAA
CAAGTCTGCC-3′ for POLH; F: 5′-GTCGTGAGAGTCGT CAGTGC-3′ and
R: 5′-GCT TGCCAGAGCGTGAAGTA-3′ for POLI; F:
5′-AGCCATGCCAGGATTTATTG-3′ and R: 5′-GGATCGTTCATGCTCACTCA-3′ for
POLK; F: 5′-AAAGAACTCCTGGAAGTGATGGA-3′ and R:
5′-GCCAAGACCCGAATGAGACC-3′ for POLQ; F:
5′-CCGTGTCCGTGGAAATCTCC-3′ and R: 5′-GTGGGGCTCTCATCTGGGAT-3′ for
REV3L; and F: 5′-TCATGGGTGTGAACCATGAGAA-3′ and R: 5′-GGCATG
GACTGTGGTCATGAG-3′ for GAPDH.
Western blot analysis
All cells were lysed with cell lysis buffer
(Beyotime, Shanghai, China) supplemented with 1 mM PMSF (Bioprimacy
Co., Ltd.). Protein extracts were separated by 10% SDS
polyacrylamide gel (Beyotime) electrophoresis and then
electrophoretically transferred to polyvinylidene fluoride
membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Following blocking with 5% skimmed milk, the membranes were
separately incubated with rabbit polyclonal antibodies against
human pol η (1:2,000), ι (1:8,000) and θ (1:1,000) (all Abcam,
Cambridge, UK), and mouse monoclonal antibodies against human pol κ
and ζ (both 1:1,000; Abcam) and GAPDH (1:1,000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) at 4°C overnight.
Secondary anti-mouse or anti-rabbit IgGs conjugated to horseradish
peroxidase (Santa Cruz Biotechnology, Inc.) were incubated with the
membranes for 1 h at room temperature at 1:8,000 dilution in PBS
containing 0.1% Tween 20 (Bioprimacy Co., Ltd.). The blots were
developed using SuperSignal West Pico chemiluminescent substrate
(Pierce Biotechnology, Inc., Rockford, IL, USA).
Viability of UV-irradiated cells
Viability of UV-irradiated cells was determined by
conducting the CellTiter-Glo luminescent cell viability assay
(Promega Corporation, Madison, WI, USA), which measures the
metabolic activity of cells by detecting cellular ATP (18).
Results
Patient characteristics
The typical signs of the sun damage phenotype were
present in the investigated patient. The patient, a farmer who had
previously had prolonged exposure to sunlight without careful
protection, separately underwent lingual and temporal tumor
resection 20 years ago. Freckling was widely distributed on the
exposed skin and recurrent skin ulcers were identified on the
patient’s hands and limbs (Fig.
1C). Squamous cell carcinoma was diagnosed on the patient’s
lower lip (Fig. 1A, B and D). In
addition, the right eye had suffered from conjunctivitis for a
number of years, which developed into glaucoma six years ago. No
neurological abnormalities were identified in the patient. The
patient has no siblings and the parents of the patient are
consanguineous and normal. Other members of the patient’s family do
not share any of these phenotypes.
PCR and sequencing of the cDNA
The genomic DNA of the patient yielded no bands
between exons 5 and 9 of the POLH gene, whereas intact bands
were observed in the DNA from the normal controls in PCR reactions
(Fig. 2A). Based on this
observation, it has been deduced that there is a large homozygous
deletion with breakpoints in introns 4 and 9 that disrupts the
intervening exons. Sequencing of the cDNA obtained from tumor cells
verified the existence of such a deletion (Fig. 3A and B). This deletion causes an
early termination at amino acid (aa) site 165 in the peptide
(Fig. 3F). Primers were designed
for introns 4 and 9 to identify the breakpoints of the deletion
(Fig. 2C; Table I). Unlike with normal cells, the
patient exhibited no bands following the region of primer 4-2 in
intron 4 and was defective within the region of primer 9-1 in
intron 9 (Fig. 2B). Thus, the
breakpoints were narrowed down to the region of primers 4-3 and
9-1. To determine the specific breakpoints, the primers 4-2 F and
9-1 R were used as the upstream and downstream primers,
respectively, to amplify the patient’s genomic DNA. A 2.8-kb band
was amplified from the DNA of the patient, but not from the DNA of
normal controls (data not shown). Sequencing of the 2.8-kb band
revealed the exact breakpoint in intron 4 to be 7306 bp from exon 4
and the breakpoint in intron 9 to be 1415 bp from exon 9 (Fig. 3C–E). The same introns of POLH
were also examined in 96 normal individuals to rule out the
possibility of this deletion being a polymorphism (data not
shown).
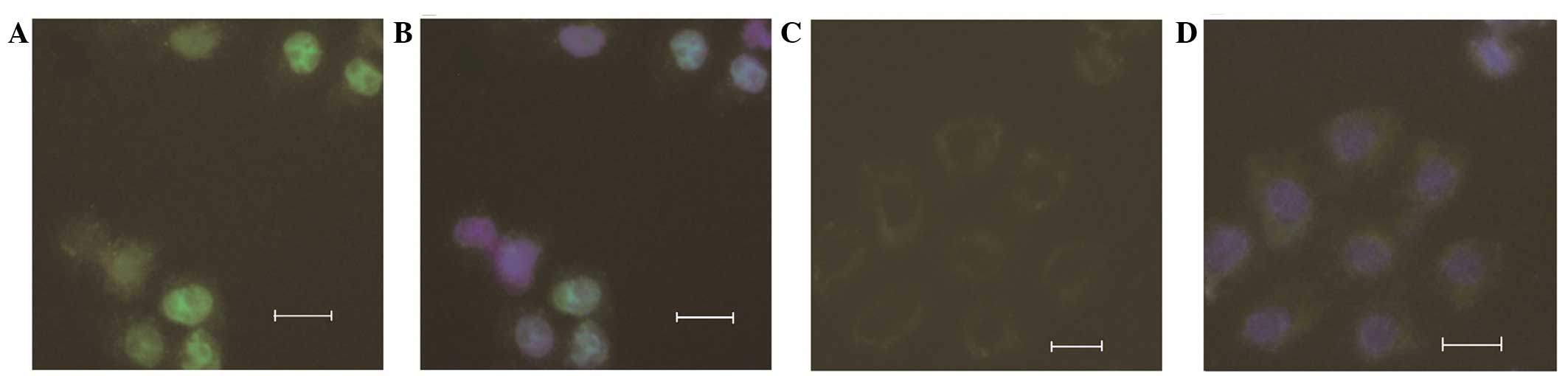
Expression of the POLH-pEYFP-C1 vector in
HeLa cells
The mutant and wild-type POLH-pEYFP-C1
vectors were transfected into HeLa cells to examine the effect of
the deletion found in the patient on the function of POLH.
Fluorescence detection showed that while wild-type pol η localized
in the cytoplasm and nucleus, the truncated pol η, encoded by
mutant POLH, did not localize in the nucleus. The intensity
of fluorescence in the nucleus was higher than that in the
cytoplasm of the wild-type POLH. In addition, the level of
YFP-tagged mutant protein was lower than that of the wild-type
protein in those cells (Fig.
4A–D).
qPCR analyses
qPCR results revealed that the mRNA quantities of
POLH, POLK, POLQ and REV3L in XP-V
tumor cells were lower than those of the controls; however, the
expression of POLI was higher in the XP-V tumor cells in the
absence of UV irradiation (P<0.05; Fig. 5A). Upon UV irradiation, all these
genes were expressed at higher levels in the tumor cells than in
the control cells (P<0.05), with the exception of REV3L,
which exhibited similar expression levels in the XP-V tumor cells
and one of the normal controls (sample UV2) (Fig. 5A). Specifically, expression of
POLK, POLQ and REV3L increased in the tumor
cells while that of POLH, POLK, POLQ and
REV3L decreased in the normal controls. Transfection of HeLa
cells with polymerase-specific siRNA effectively knocked down the
mRNA levels of POLH (P<0.01), and expression of all but
one (POLI) of the tested genes was found to decrease in
these cells (P<0.05; Fig. 5B).
Detailed data are presented in Table
II.
 | Figure 5Results of qPCR analyses of
POLH, POLI, POLK, POLQ and REV3L
in control and UV-exposed cells. (A) Sample P, XP-V tumor cells
from the patient; samples 1–3, normal cells from three controls;
and samples UVP, UV1, UV2 and UV3, samples P, 1, 2 and 3 that
underwent UV irradiation, respectively. Prior to UV irradiation,
lower expression of POLH, POLK, POLQ and
REV3L and higher expression of POLI was observed in
XP-V tumor cells (sample P) compared with normal controls (samples
1–3) (P<0.05). Following UV irradiation, expression of
POLK, POLQ and REV3L was found to have
significantly increased in XP-V tumor cells. All other genes, with
the exception of REV3L, had higher expression in tumor cells
(sample UVP) than in normal cells (samples UV1-UV3) (P<0.05).
(B) Tumor samples (sample P) and HeLa cells were set as reference
samples and the RNA quantity of each tested gene was defined as
1.0. Prior to UV irradiation, POLH, POLK, POLQ
and REV3L had lower expression in the SiHeLa sample than in
the HeLa sample (P<0.05). Following UV irradiation, all tested
genes had lower expression in the SiUVHeLa sample than in the
UVHeLa sample (P<0.05). *Statistically significant
difference between samples P and 1, 2 and 3, and between SiHeLa and
HeLa cells; *statistically significant difference
between samples UVP and UV1, UV2 and UV3, and between SiUVHeLa and
UVHeLa cells. One symbol indicates P<0.05 and two symbols
indicates P<0.01. qPCR, real-time polymerase chain reaction;
XP-V, xeroderma pigmentosum variant; RQ, mRNA relative quantity;
HeLa, HeLa cells without POLH knockdown; SiHeLa, HeLa cells
with POLH knockdown; SiUVHeLa, UV-irradiated HeLa cells with
POLH knockdown; UVHeLa; UV-irradiated HeLa cells without
POLH knockdown. |
| Table IIDetailed statistics of qPCR
results. |
Table II
Detailed statistics of qPCR
results.
| Group A | Group B | Group UVA | Group UVB |
|---|
|
|
|
|
|
|---|
| Gene | Sample P | SiHeLa | Sample 1 | Sample 2 | Sample 3 | HeLa | Sample UVP | SiUVHeLa | Sample UV1 | Sample UV2 | Sample UV3 | UVHeLa |
|---|
| POLH | 1±0.12 | 0.40±0.06 | 1.81±0.18a,c | 3.34±0.19a,c | 2.90±0.21a,c | 1±0.10a,c | 1.23±0.06 | 0.26±0.04 | 0.76±0.09b,c | 0.84±0.08b,c | 0.87±0.09b,c | 0.530±0.05b,c |
| POLI | 1±0.17 | 1.00±0.13 | 0.44±0.05a,c | 0.35±0.04a,c | 0.61±0.09a,d | 1±0.10 | 0.73±0.03 | 0.43±0.14 | 0.32±0.07b,c | 0.42±0.10b,d | 0.58±0.03b,c | 0.825±0.07b,d |
| POLK | 1±0.15 | 0.66±0.09 | 10.95±0.85a,c | 10.79±1.04a,c | 12.66±0.70a,c | 1±0.14a,d | 10.20±0.91 | 0.25±0.14 | 5.75±0.95b,c | 6.88±0.96b,d | 4.98±0.30b,c | 0.700±0.13b,d |
| POLQ | 1±0.15 | 0.63±0.06 | 5.67±0.48a,c | 10.52±0.65a,c | 7.71±0.48a,c | 1±0.10a,c | 12.6±0.65 | 0.32±0.04 | 4.91±0.57b,c | 6.79±0.83b,c | 2.58±0.33b,c | 0.810±0.08b,c |
| REV3L | 1±0.14 | 0.68±0.06 | 8.39±0.58a,c | 5.86±0.75a,c | 7.78±0.63a,c | 1±0.12a,d | 4.97±0.25 | 0.19±0.10 | 2.95±0.41b,c | 4.65±0.35 | 2.73±0.20b,c | 0.640±0.12b,c |
Western blot analyses
Concordant with the decrease in the mRNA quantities
of pol κ, θ and ζ, their protein levels were also found to
significantly decrease in the XP-V tumor cells compared with those
of the controls. Following UV irradiation, pol κ and θ were found
to be at higher levels in the XP-V tumor cells, whereas pol ι and ζ
were at comparable levels in the XP-V tumor cells and normal
controls (Fig. 6A). XP-V cell lines
(XP1SF and XP30RO) expressing mutant pol η verified these changes
when the cell lines were compared with the HSF cell line (Fig. 6B). As the defective pol η lacked the
motif normally recognized by its antibody, binding of pol η was not
observed in the western blot assays. Similar to the results of the
qPCR analyses, HeLa cells subjected to UV irradiation exhibited
lower levels of all the tested proteins when POLH was
knocked down (Fig. 6C).
 | Figure 6Western blot analysis of pol η, ι, κ,
θ and ζ. GAPDH protein levels were used for normalization. (A)
Tested polymerases in tumor cells from the XP-V patient and normal
cells from three controls, including samples P, tumor cells from
the patient; 1–3, normal cells from three controls; and RP, R1, R2
and R3, cells from samples P, 1, 2 and 3 that underwent UV
irradiation, respectively. (B) Tested polymerases in XP-V and HSF
cell lines, including samples X1 and X2, cells from XP30RO and
XP1SF, respectively; SF, cells from HSF cell line; and RX1, RX2 and
RSF, cells from samples X1, X2 and SF that underwent UV
irradiation, respectively. (C) Tested polymerases in HeLa cells,
including samples Si, HeLa cells with POLH knockdown; NSi,
HeLa cells without POLH knockdown; and RSi and RNSi, cells
from samples Si and NSi, respectively, that underwent UV
irradiation. XP-V, xeroderma pigmentosum variant; pol η, polymerase
η. |
Cell viability
The qPCR and western blot analyses revealed that, in
the epithelial control, HeLa and HSF cells, expression of all the
tested genes was found to decrease following UV irradiation. We
hypothesized that this may have been caused by the reduction of
cell viability following UV irradiation. Cellular ATP levels were
measured to assay the activity of these cells. The viability of the
cells was observed to decrease substantially (52±5% in normal
epithelial, 47±6% in HSF and 36±4% in HeLa cells) 48 h following UV
irradiation, therefore confirming the hypothesis.
Discussion
Pol η is an essential polymerase that allows cells
to bypass a DNA lesion during DNA replication. Although pol η
usually shows low fidelity (22) in
DNA replication, it replaces conventional polymerases with high
fidelity to easily perform TLS in the replication of photodamaged
DNA (23). Pol η is the only
error-free polymerase reported to bypass the thymine-thymine CPD, a
UV-induced DNA damage product (4,23). In
XP-V patients with POLH mutations, when skin cells have
suffered UV irradiation and generate harmful products, the
defective pol η does not bypass CPD broken sites and therefore, DNA
replication does not continue normally (4,24). As
a result, UV lesions are highly mutagenic, leading to skin
cancer.
Human POLH is located on chromosome 6p21.1
and encodes a peptide of 713 aa. Previous studies have demonstrated
that the N-terminal, 511 aa of the polymerase, is necessary for its
activity (25) while the
C-terminal, 70 aa, is responsible for its nuclear localization and
a further 50 aa are required for relocalization following UV
irradiation (10). Although a
number of missense and small deletion mutations have been found in
the POLH gene, no previous studies have reported a large
deletion spanning greater than one exon in this gene. The present
study revealed a large deletion in the POLH gene that
disrupts the region between exons 5 and 9. This mutation is likely
to result in the severe truncation of pol η, with only 164 aa
remaining intact and the loss of key regions responsible for
activity and nuclear localization (Fig.
3F). Consistent with this hypothesis and previous observations,
the mutation was found to lead to alteration in the cellular
localization of the protein.
Of note, the XP-V patient investigated in the
present study survived longer than the majority of XP patients
(26). Although the patient
exhibited a severe truncation of the POLH gene, the patient
did not exhibit more dangerous phenotypes or have a shorter
lifespan than other XP-V cases (8).
It has been proposed that the phenotype of XP patients depends on
their protection against sun toxicity, but not the mutation
(2). However, the subject of the
present study was a farmer who had been exposed to prolonged
sunlight without careful protection, but did not show more severe
phenotypes. It has also been reported that XP-V patients often
possess relatively mild phenotypes and rarely develop neurological
abnormalities compared with other XP patients (2). Therefore, the factors avoiding severe
damage in XP-V patients requires further investigation.
Previous studies have demonstrated that POLI,
POLK and REV3L are responsible for CPD TLS (18,27,28).
Such genetic instability is caused by the combined effect of
several unusual polymerases. Knockdown of REV3L alone,
POLK and POLI together, or POLK and
REV3L together, has been previously reported to lead to a
significant decrease in accurate TLS of XP-V cells (18). In the present study, low expression
of POLH, POLK and REV3L was observed in the
XP-V tumor cells. This indicates that the low efficiency of TLS
contributes to genetic instability in XP-V tumor cells and involves
the imbalance of other specific polymerases besides defective pol
η. As pol η is the only error-free polymerase in the TLS of CPDs,
when it loses its function, other error-prone polymerases may
maintain their low expression to avoid mismatch in the replication
of CPDs. POLI, the only gene with a higher expression in
XP-V tumor cells, may compensate for TLS. Consistent with this, low
expression of pol κ, ζ and θ was found in the XP-V cell lines and
HeLa cells with POLH knockdown. As the XP-V tumor often
occurs in exposed regions, it is possible that the damaged cells
have been exposed to high levels of UV. Considering that the
patient in the current study was a farmer with potentially high
exposure to irradiation, we hypothesized that this may have been
the cause of XP disease. In addition, increased expression of pol κ
and ζ was observed in the XP-V cells following strong UV
irradiation. When DNA has been damaged by UV irradiation and pol η
completely loses its function, pol κ and ζ may compensate for TLS,
which may explain the higher expression of these genes in the XP-V
tumor cells and cell lines. In the current study, following UV
irradiation, all tested genes were found to have a lower expression
in the HeLa cells with POLH knockdown. This may be due to
residual pol η, which may function normally in the TLS of CPDs,
leading other genes to maintain a low expression to avoid mismatch
in the replication of CPDs.
Based on these observations, we hypothesized that
when the XP-V cells suffer strong UV toxicity and generate numerous
photodimers, they synthesize more pol κ and ζ to compensate for the
defective pol η. However, pol κ and ζ are error-prone, therefore,
these polymerases promote mismatch in DNA replication and
accelerate the genetic instability (18), which may facilitate tumor formation.
If patients avoid UV irradiation, it is likely that the skin cells
maintain a low level of pol κ and ζ to prevent error-prone DNA
replication. However, low levels of pol η, κ and ζ decrease the
efficiency of TLS, and therefore, DNA lesion replication is
disrupted as well. It has been previously demonstrated that the
upregulation of pol θ perturbs DNA replication, promotes genetic
instability and is associated with poor prognosis in breast cancer
(29). The significant increase in
the expression levels of pol θ observed in the XP-V tumor cells and
cell lines following UV irradiation in the present study may also
indicate genetic instability.
In summary, the current study reported a novel,
large deletion of POLH in a XP-V patient. qPCR and western
blot analyses of cells expressing mutant POLH were
conducted. The results indicated that genetic instability in XP-V
tumors may arise due to imbalances in DNA polymerases, which may be
contributed not only by defects in pol η, but also by the unusual
expression of other polymerases. Further investigation is required
to clarify the correlation between genotype and resulting phenotype
in XP-V, as well as to elucidate the molecular mechanism involved
in XP-V tumor formation.
Acknowledgements
The authors would like to thank the patient and
other individuals for participating in the present study. The study
was supported by grants from the National Natural Science
Foundation of China (grant nos. 30930099, 81120108010 and
81170957).
References
|
1
|
Kraemer KH, Lee MM and Scotto J: Xeroderma
pigmentosum. Cutaneous, ocular and neurologic abnormalities in 830
published cases. Arch Dermatol. 123:241–250. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lehmann AR, McGibbon D and Stefanini M:
Xeroderma pigmentosum. Orphanet J Rare Dis. 6:702011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kraemer KH and DiGiovanna JJ: Xeroderma
Pigmentosum. GeneReviews. Pagon RA, Bird TD, Dolan CR and Stephens
K: Seattle, WA: pp. 1993–2013. 2003
|
|
4
|
Masutani C, Kusumoto R, Yamada A, et al:
The XPV (xeroderma pigmentosum variant) gene encodes human DNA
polymerase eta. Nature. 399:700–704. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johnson RE, Kondratick CM, Prakash S and
Prakash L: hRAD30 mutations in the variant form of xeroderma
pigmentosum. Science. 285:263–265. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inui H, Oh KS, Nadem C, et al: Xeroderma
pigmentosum-variant patients from America, Europe and Asia. J
Invest Dermatol. 128:2055–2068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gratchev A, Strein P, Utikal J and Sergij
G: Molecular genetics of Xeroderma pigmentosum variant. Exp
Dermatol. 12:529–536. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Broughton BC, Cordonnier A, Kleijer WJ, et
al: Molecular analysis of mutations in DNA polymerase eta in
xeroderma pigmentosum-variant patients. Proc Natl Acad Sci USA.
99:815–820. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Faili A, Stary A, Delbos F, et al: A
backup role of DNA polymerase kappa in Ig gene hypermutation only
takes place in the complete absence of DNA polymerase eta. J
Immunol. 182:6353–6359. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kannouche P, Broughton BC, Volker M,
Hanaoka F, Mullenders LH and Lehmann AR: Domain structure,
localization and function of DNA polymerase eta, defective in
xeroderma pigmentosum variant cells. Genes Dev. 15:158–172. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yuasa M, Masutani C, Eki T and Hanaoka F:
Genomic structure, chromosomal localization and identification of
mutations in the xeroderma pigmentosum variant (XPV) gene.
Oncogene. 19:4721–4728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tanioka M, Masaki T, Ono R, et al:
Molecular analysis of DNA polymerase eta gene in Japanese patients
diagnosed as xeroderma pigmentosum variant type. J Invest Dermatol.
127:1745–1751. 2007.PubMed/NCBI
|
|
13
|
Hentosh P, Benjamin T, Hall L, et al:
Xeroderma pigmentosum variant: complementary molecular approaches
to detect a 13 base pair deletion in the DNA polymerase eta gene.
Exp Mol Pathol. 91:528–533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Johnson RE, Prakash S and Prakash L:
Efficient bypass of a thymine-thymine dimer by yeast DNA
polymerase, Poleta. Science. 283:1001–1004. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ohmori H, Friedberg EC, Fuchs RP, et al:
The Y-family of DNA polymerases. Mol Cell. 8:7–8. 2001. View Article : Google Scholar
|
|
16
|
Daya-Grosjean L and Sarasin A: The role of
UV induced lesions in skin carcinogenesis: an overview of oncogene
and tumor suppressor gene modifications in xeroderma pigmentosum
skin tumors. Mutat Res. 571:43–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Silverstein TD, Johnson RE, Jain R, et al:
Structural basis for the suppression of skin cancers by DNA
polymerase eta. Nature. 465:1039–1043. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ziv O, Geacintov N, Nakajima S, Yasui A
and Livneh Z: DNA polymerase zeta cooperates with polymerases kappa
and iota in translesion DNA synthesis across pyrimidine photodimers
in cells from XPV patients. Proc Natl Acad Sci USA.
106:11552–11557. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Masuda K, Ouchida R, Hikida M, et al: DNA
polymerases eta and theta function in the same genetic pathway to
generate mutations at A/T during somatic hypermutation of Ig genes.
J Biol Chem. 282:17387–17394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song Y, Wang C, Peng B, et al: Phenotypes
and genotypes in 2 DGI families with different DSPP mutations. Oral
Surg Oral Med Oral Pathol Oral Radiol Endod. 102:360–374. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
22
|
Johnson RE, Washington MT, Prakash S and
Prakash L: Fidelity of human DNA polymerase eta. J Biol Chem.
275:7447–7450. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cruet-Hennequart S, Gallagher K, Sokol AM,
Villalan S, Prendergast AM and Carty MP: DNA polymerase eta, a key
protein in translesion synthesis in human cells. Subcell Biochem.
50:189–209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Svoboda DL, Briley LP and Vos JM:
Defective bypass replication of a leading strand cyclobutane
thymine dimer in xeroderma pigmentosum variant cell extracts.
Cancer Res. 58:2445–2448. 1998.
|
|
25
|
Masutani C, Araki M, Yamada A, et al:
Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa
cells has a thymine dimer bypass DNA polymerase activity. EMBO J.
18:3491–3501. 1999. View Article : Google Scholar
|
|
26
|
Bradford PT, Goldstein AM, Tamura D, et
al: Cancer and neurologic degeneration in xeroderma pigmentosum:
long term follow-up characterises the role of DNA repair. J Med
Genet. 48:168–176. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gueranger Q, Stary A, Aoufouchi S, et al:
Role of DNA polymerases eta, iota and zeta in UV resistance and
UV-induced mutagenesis in a human cell line. DNA Repair (Amst).
7:1551–1562. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Woodgate R, McManus TP, et al:
Evidence that in xeroderma pigmentosum variant cells, which lack
DNA polymerase eta, DNA polymerase iota causes the very high
frequency and unique spectrum of UV-induced mutations. Cancer Res.
67:3018–3026. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lemée F, Bergoglio V, Fernandez-Vidal A,
et al: DNA polymerase theta up-regulation is associated with poor
survival in breast cancer, perturbs DNA replication and promotes
genetic instability. Proc Natl Acad Sci USA. 107:13390–13395.
2010.PubMed/NCBI
|