Introduction
Aplastic anemia (AA) is a clinical syndrome of
peripheral blood pancytopenia and a hypocellular bone marrow.
Immunosuppressive therapy is a key treatment strategy for AA.
Genomic instability in AA does not appear to be a rare event. AA
evolves into acute myeloid leukemia (AML) in 5–15% of all cases
(1,2). Careful observation for leukemic
transformation is indicated in patients with severe AA (SAA).
Trisomy 21 is relatively common in Down syndrome, but rare in
secondary AML. The current report presents the first case of SAA
preceding acute monocytic leukemia (AML-M5) with acquired trisomy
21. Written informed consent was obtained from the patient’s
family.
Case report
A 20-year-old female was admitted to the Department
of Hematology, The Central Hospital of Taian (Taian, China) in May
1996 with fever, dizziness and increasing tiredness. The patient
exhibited no other positive medical or family history of any
specific disease. In addition, the patient did not exhibit any
features of Down syndrome. On physical examination, the patient was
severely ill, pale and covered with mucocutaneous petechial
bleeding. No lymphadenopathy or hepatosplenomegaly was observed.
Viral serology was negative for hepatitis A, B and C, as well as
human immunodeficiency virus. On full blood count, hemoglobin (Hb)
count was 78 g/l, white blood cell count was 0.7×109/l,
platelet count was 16×109/l, neutrophil count was
0.46×109/l and reticulocyte count was 0.6%. Bone marrow
aspirate was profoundly hypocellular and no megakaryocytes were
observed. No mast cells, but a few foci of plasma cells and
lymphocytes were identified. Residual myeloid and erythroid
precursors were observed without any abnormal infiltrate. The
patient was diagnosed with SAA.
The individual was treated with intravenous
broad-spectrum antibiotics and multiple red cell and platelet
transfusions with resolution of the fever, dizziness and increasing
tiredness. The patient received, with high-dose methylprednisolone,
cyclosporine A, androgens and recombinant human granulocyte
colony-stimulating factor (G-CSF). Two years later, the blood
counts returned to normal and the patient gradually stopped
receiving drug therapy.
In 2001, the patient delivered a normal child.
However, the results of the patients routine blood test were
abnormal; the platelet count was <10×109/l and Hb
levels were also decreased. The patient received blood and platelet
transfusions without other drug treatment.
In October 2010, the patient was hospitalized due to
nasal bleeding and mild headache. On full blood count, white blood
cell count was 19.92×109/l, neutrophil count was
5.89×109/l, monocyte count was 2.09×109/l, Hb
count was 105 g/l, platelet count was 20×109/l and the
reticulocyte count was 4.27%. Blast cells were observed on blood
film and the bone marrow smears showed an increased number (31%) of
monoblasts and promonocytes. Blast cells stained negatively for PAS
and specific esterase stain and positive for non-specific esterase,
which was inhibited by sodium fluoride. These observations were
compatible with AML-M5b and chromosomal examination revealed
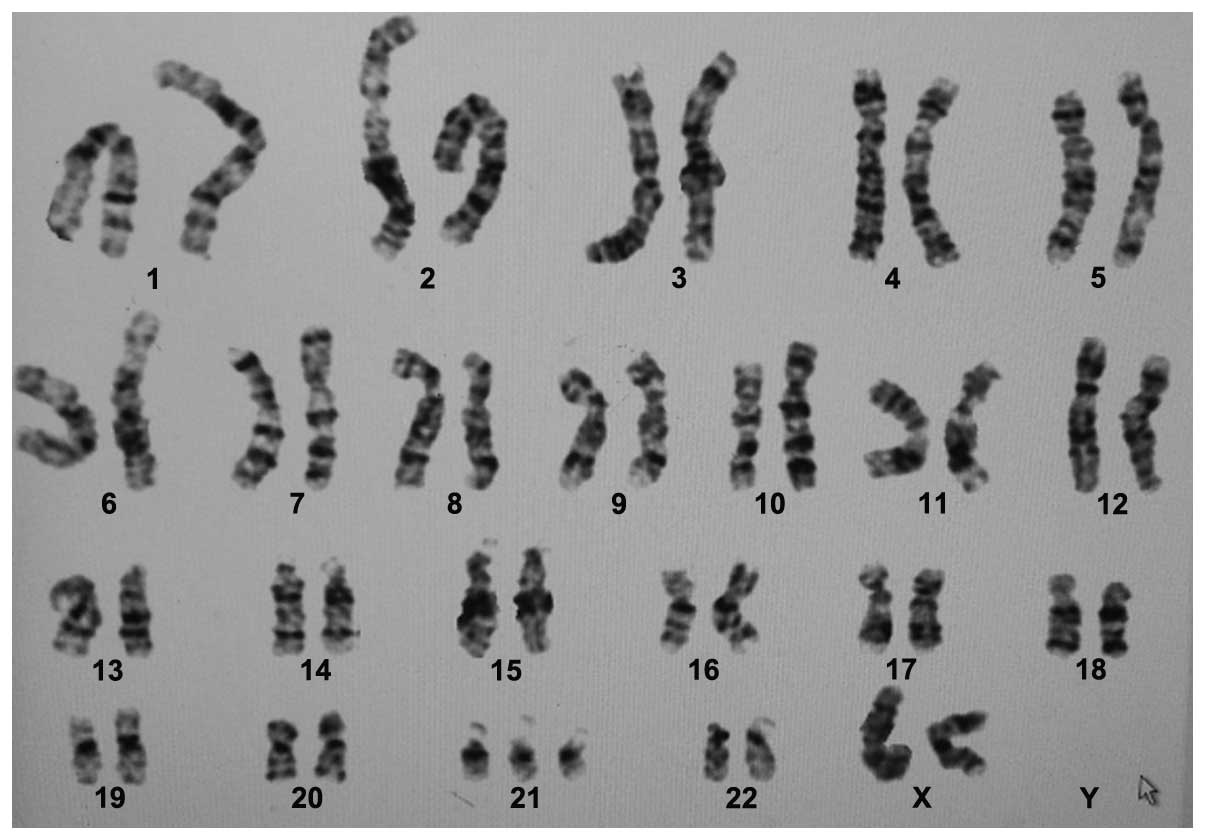
47,XX,+21[10]/46,XX[1] (Fig. 1).
The patient was treated with induction chemotherapy, but did not
achieve remission. The patient succumbed to central nervous system
bleeding 2 weeks following chemotherapy.
Discussion
Leukemia is a rare and late complication of AA. AML
and myelodysplasia syndromes (MDS) occur in 15% of patients within
10 years following immunosuppressive treatment (3). There have been various reasons
proposed as to why AA may precede AML. Previously, Barrios et
al(4) presented a patient with
SAA, with no previous chromosomal abnormalities, who developed
trisomy 21 and monosomy 7 during treatment with intravenous
cyclosporine. The abnormal karyotype disappeared when the drug was
changed to the oral form. Furthermore, the published data on the
safety of G-CSF for AA remain controversial. It has been reported
that G-CSF therapy does not increase the risk of t-MDS/AML
development (5–7). However, specific results have shown
that G-CSF therapy is one of the risk factors for AA evolution to
MDS/AML (8–10). Further studies are required to
identify the risk factors in SAA for developing MDS/AML.
The evolution of AA to clonal hematologic diseases
is well recognized. Cytogenetics are usually normal in the aplastic
phase, but abnormalities may develop in the leukemic phase. The
current report presents a new case of SAA preceding AML with
trisomy 21 as the sole acquired karyotypic abnormality.
Trisomy 21 has been demonstrated to be a recurring
cytogenetic abnormality in AML and MDS. Trisomy may contribute to
leukemogenesis by a gene dosage effect. The majority of adult AML
cases with trisomy 21 have been associated with AML-M2 or -M4
(11). By comparison, the most
common hematological malignancy in patients with Down syndrome is
AML-M7 (8,12). The prognostic significance of
acquired trisomy 21 as the sole abnormality in adult AML remains
unclear. A higher complete remission has previously been reported
in AML with trisomy 21 (13), but a
study showed that other accompanied cytogenetic changes determined
the clinical outcome (14). In
addition, a greater number of studies have shown that AML patients
with acquired trisomy 21 as the sole abnormality exhibit a poor
prognosis (14–16). Therefore, further studies with
larger cohorts of patients are required to evaluate the prognostic
significance of acquired trisomy 21.
The course and prognosis of secondary leukemia
depends on not only cytogenetic features, but also clinical and
molecular features at diagnosis. Previously, the successful therapy
of such secondary leukemia has been rarely reported. Patients have
not responded or have succumbed to infection or bleeding during
induction. Irreversible aplasia is a hazard of intensive
chemotherapy. Fatal bleeding events eventually occurred in the
present patient, who was refractory to platelet transfusions.
Cytomorphological and cytogenetic abnormalities are
rarely observed in AA, which may predict patients with SAA at risk
for leukemia. Prior to the diagnosis of AML, SAA patients may
develop signs of MDS. Therefore, long-term follow-up is essential
to assess the incidence and risk factors for evolution of AA into
AML, and to administer salvage therapy for transformation, in time,
during follow-up. In addition, the best supportive care must be
fully integrated with diagnosis and treatment. For example,
multiple anti-human leukocyte antigen (HLA) antibodies must be
detected prior to the initiation of chemotherapy and appropriate,
unrelated HLA-matched platelet donors must be selected prior to
therapy in patients who were refractory to platelet transfusions
(17). In conclusion, an increased
number of factors must be considered when determining the most
appropriate management of such secondary leukemia. This remains an
unsatisfactory area with the greatest clinical challenge in
secondary AML.
Acknowledgements
The present study was supported by grants from the
Shandong Province Natural Science Foundation of China (nos.
2009ZRA09006 and ZR2012HL38) and the Shandong Province Medical
Science and Technology Development Program (no. 2011HW077).
References
|
1
|
Ohara A, Kojima S, Hamajima N, et al:
Myelodysplastic syndrome and acute myelogenous leukemia as a late
clonal complication in children with acquired aplastic anemia.
Blood. 90:1009–10013. 1997.PubMed/NCBI
|
|
2
|
Barrett J, Saunthararajah Y and Molldrem
J: Myelodysplastic syndrome and aplastic anemia: distinct entities
or diseases linked by a common pathophysiology? Semin Hematol.
37:15–29. 2000. View Article : Google Scholar
|
|
3
|
Tichelli A, Gratwohl A, Nissen C and Speck
B: Late clonal complications in severe aplastic anemia. Leuk
Lymphoma. 12:167–175. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barrios NJ, Kirkpatrick DV, Levin ML and
Varela M: Transient expression of trisomy 21 and monosomy 7
following cyclosporin A in a patient with aplastic anemia. Leuk
Res. 15:531–533. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Locasciulli A, Arcese W, Locatelli F, Di
Bona E and Bacigalupo A; Italian Aplastic Anaemia Study Group.
Treatment of aplastic anaemia with granulocyte-colony stimulating
factor and risk of malignancy: Italian Aplastic Anaemia Study
Group. Lancet. 357:43–44. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Imashuku S, Hibi S, Bessho F, et al:
Detection of myelodysplastic syndrome/acute myeloid leukemia
evolving from aplastic anemia in children, treated with recombinant
human G-CSF. Haematologica. 88:ECR312003.
|
|
7
|
Gurion R, Gafter-Gvili A, Paul M, et al:
Hematopoietic growth factors in aplastic anemia patients treated
with immunosuppressive therapy - systematic review and
meta-analysis. Haematologica. 94:712–719. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kojima S, Ohara A, Tsuchida M, et al: Risk
factors for evolution of acquired aplastic anemia into
myelodysplastic syndrome and acute myeloid leukemia after
immunosuppressive therapy in children. Blood. 100:786–790. 2002.
View Article : Google Scholar
|
|
9
|
Sloand EM, Yong AS, Ramkissoon S, et al:
Granulocyte colony-stimulating factor preferentially stimulates
proliferation of monosomy 7 cells bearing the isoform IV receptor.
Proc Natl Acad Sci USA. 103:14483–14488. 2006. View Article : Google Scholar
|
|
10
|
Socie G, Mary JY, Schrezenmeier H, Marsh
J, et al: Granulocyte-stimulating factor and severe aplastic
anemia: a survey by the European Group for Blood and Marrow
Transplantation (EBMT). Blood. 109:2794–2796. 2007.
|
|
11
|
Wang HF, Cheng YZ, Wang HP, Chen ZM, Lou
JY and Jin J: CD19-positive acute myeloblastic leukemia with
trisomy 21 as a sole acquired karyotypic abnormality. J Zhejiang
Univ Sci B. 10:833–838. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Langebrake C, Creutzig U and Reinhardt D:
Immunophenotype of Down syndrome acute myeloid leukemia and
transient myeloproliferative disease differs significantly from
other diseases with morphologically identical or similar blasts.
Klin Padiatr. 217:126–134. 2005. View Article : Google Scholar
|
|
13
|
Udayakumar AM, Pathare AV, Muralitharan S,
Alghzaly AA, Alkindi S and Raeburn JA: Trisomy 21 as a sole
acquired abnormality in an adult Omani patient with CD7- and
CD9-positive acute myeloid leukemia. Arch Med Res. 38:797–802.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cortes JE, Kantarjian H, O’Brien S, et al:
Clinical and prognostic significance of trisomy 21 in adult
patients with acute myelogenous leukemia and myelodysplastic
syndromes. Leukemia. 9:115–117. 1995.PubMed/NCBI
|
|
15
|
Wan TS, Au WY, Chan JC, Chan LC and Ma SK:
Trisomy 21 as the sole acquired karyotypic abnormality in acute
myeloid leukemia and myelodysplastic syndrome. Leuk Res.
23:1079–1083. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Ni W, Chen Z, et al: Clinical and
cytogenetic features of hematologic malignancies associated with
acquired trisomy 21. Zhonghua Yi Xue Yi Chuan Xue Za Zhi.
25:576–578. 2008.(In Chinese).
|
|
17
|
Xia WJ, Ye X, Tian LW, et al:
Establishment of platelet donor registry improves the treatment of
platelet transfusion refractoriness in Guangzhou region of China.
Transfus Med. 20:269–274. 2010. View Article : Google Scholar
|