1. Introduction
Interleukin (IL)-9 is a member of the common γ-chain
family of cytokines, using the γ-chain receptor in combination with
the cytokine-specific receptor, IL-9 receptor (IL-9R) α (1). There has been renewed interest in IL-9
since the identification of a subset of T cells which produce this
cytokine. However, previous conflicting studies have been
identified, concerning which T cells produce this cytokine. The
studies demonstrated, but are not limited to, the following T
cells: T helper (Th) 2, 9 and 17 cells and regulatory T (Treg)
cells. Besides the role of IL-9 during immune responses, its growth
factor and antiapoptotic activities on multiple transformed cells
suggest a potential role in hematological malignancies. Notably,
IL-9 overexpression induces thymic lymphomas in mice, and IL-9
production has an effect on Hodgkin’s disease and human
T-lymphotropic virus type I (HTLV-I)-transformed T cells in humans.
IL-9 activities also involve IL-2, -4, -7, -15 and -21 signaling,
which is mediated by a specific receptor chain that forms a
heterodimeric receptor with the common γ chain (2). The IL-9R and common γ chains associate
with Janus kinase (JAK) 1 and JAK3 and trigger the signal
transducer and activator of transcription (STAT)-1, -3 and -5,
insulin receptor signaling (IRS) and RAS-mitogen-activated protein
kinase (MAPK) pathways. In addition, IL-9 is not expressed by Th 2
and 9 cells in the absence of STAT6 expression. Dysregulated IL-9
response also leads to autonomous cell growth and the malignant
transformation of lymphoid cells associated with constitutive
activation of the JAK/STAT pathway in vitro. The current
review summarizes the characterization of the biological activities
of IL-9, highlights the clearly defined roles of the cytokine, and
outlines questions with regard to the functions of IL-9 that
require further exploration and their downstream signaling
proteins, STATs.
2. IL-9 production and function
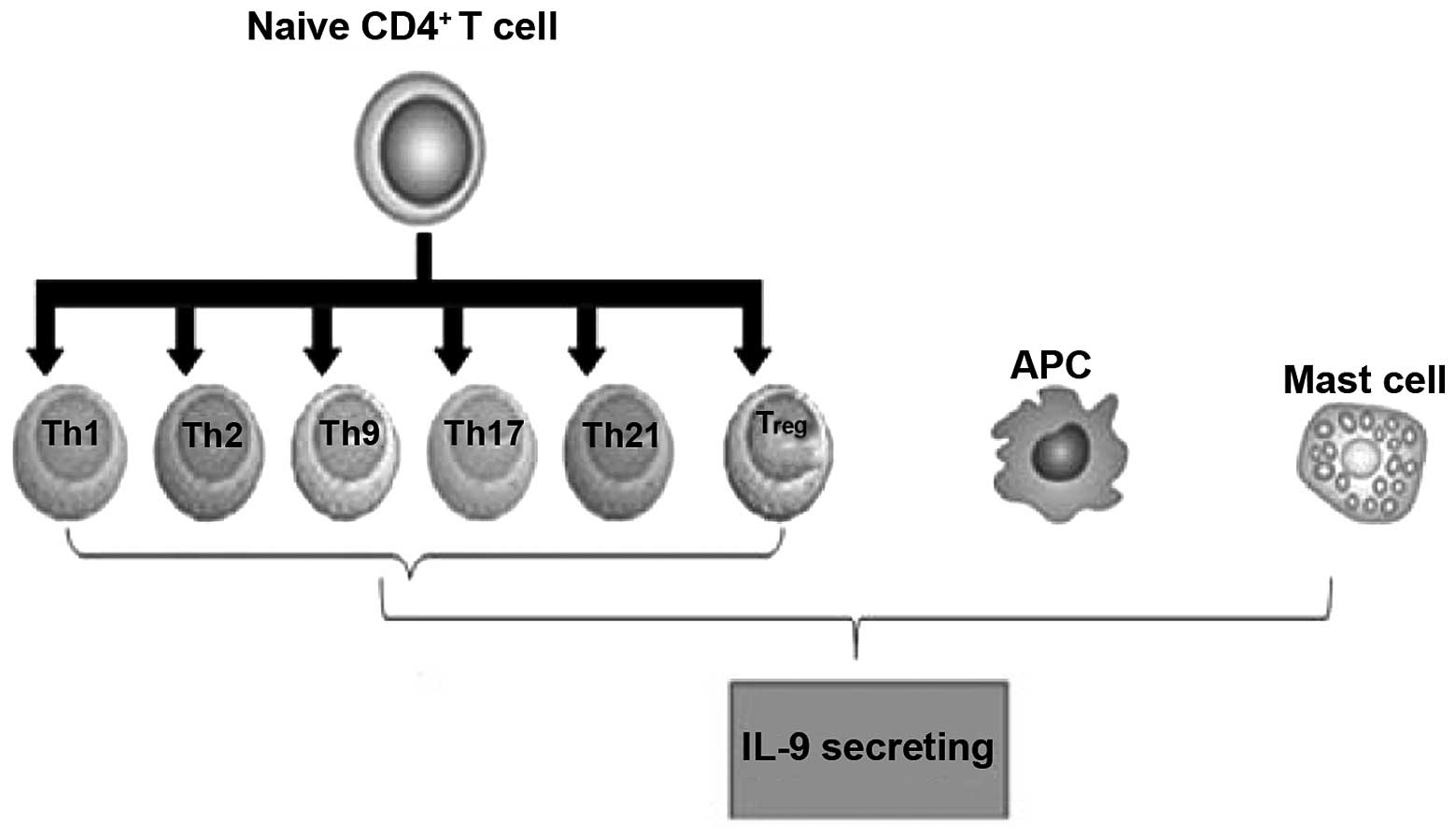
IL-9-secreting cells
Initially, IL-9 was described as a T cell-derived
cytokine with pleiotropic activities on various cell types. IL-9 is
mainly expressed by activated CD4+ T cells, including
Th2, Th9, Th17 and Treg cells (3).
As compared with plate-bound anti-CD3 mAb/soluble anti-CD28 mAb
plus transforming growth factor (TGF)-β stimulation, IL-4 and TGF-β
stimulate memory CD4(+), CD25(−) and CD45RO(+) T cell expression,
inducing higher levels of IL-9 expression, but reducing forkhead
box (Fox) p3 protein expression. IL-4 and TGF-β inhibit the
expression of Foxp3 that induces Treg generation and promotes an
IL-9-secreting phenotype, which is dependent on STAT6. Similarly,
Gata3 is required for the generation of IL-9-secreting cells,
suggesting that the IL-9-secreting population has shared factors in
their development. Human T cells also acquire IL-9-secreting
potential when cultured with TGF-β and IL-4. IL-9 production was
first associated with the Th2 phenotype (4). However, it was unclear whether
specialized cells were responsible for IL-9 production and
secretion, as was being established at the time for other
cytokines, including IL-4 and interferon (IFN) γ, in Th2 and Th1
cells, respectively.
Studies have shown that two transcription factors
are required for IL-9-secreting cells and have been reported to
bind directly to the Il9 gene. PU.1 is an ETS-family
transcription factor. PU.1-deficient T cells exhibit diminished
IL-9 production and ectopic expression of PU.1 increases IL-9
production from Th2 or Th9 cultures (5–7). PU.1
binds directly to the Il9 gene and histone modifications
associated with the Th9 phenotype are dependent upon PU.1.
PU.1-specific small interfering RNA results in impaired IL-9
production by human T cells; therefore, PU.1 is also important for
IL-9 production in human T cells. Notably, PU.1 is expressed in
greater amounts in cells cultured under Th9 conditions compared
with Th2 cells, which suggests that PU.1 is a critical factor in
diverting Th2 cells into an IL-9-secreting lineage (7). In a previous follow-up study, in the
absence of PU.1, a decreased association between Gcn5 and
p300/CREB-binding protein (CBP) associated factor and inhibition of
the expression of Gcn5 was found to result in reduced IL-9
production, which suggested that Gcn5 may be important in
PU.1-dependent IL-9 production. Similar to PU.1, IFN-regulatory
factor 4 (IRF4) was also shown to be required for Th9 generation
and, possibly in concert with PU.1, since IRF4 was originally
identified as a PU.1-interacting protein, IRF4 binds to the
Il9 gene directly. In addition, IRF4 expression has been
associated with human and mouse Th9 differentiation and is induced
by cytokines that promote IL-9 production, including IL-4, IL-2 and
TGF-β (8). IRF4 is important in the
cytokine-secreting potential of several Th subsets as IRF4 is also
required for Th2 and Th17 development (9–11).
Mast cells (MCs) also produce IL-9 in response to
lipopolysaccharides and IL-1, which are associated with the
presence of nuclear factor κB (NF-κB) binding sites in the
Il9 promoter that mediate gene activation (12–14).
Gata1 in MCs promotes IL-9 production and Il9 promoter
activation is dependent upon p38 MAPK (15). MCs and T cells, as well as the
scenarios in which each cell may contribute to IL-9 production
in vivo, have not previously been determined. In addition,
IL-9 induces MC production of TGF-β, which exhibits proinflammatory
downstream effects (Fig. 1).
IL-9R signaling and expression
The functions of IL-9 are mediated by the IL-9R,
which is a member of the hematopoietin receptor superfamily
(16). The IL-9R is shared with the
IL-2, -4, -7, -15 and -21 receptors, including the ligand-specific
α chain and the common γ chain. The mouse receptor contains 468
amino acids, but the human IL-9R gene contains 11 exons and encodes
a 522-amino acid protein. On the basis of the presence of the WSXWS
motif in the extracellular domain and Box1 and Box2 motifs in the
intracellular domain, the IL-9Rα is a member of the hematopoietin
superfamily. The Box1 intracellular domain is critical for
IL-9-induced cell growth, activation of STAT3 and induced gene
expression, which is between amino acids 338 and 422, including a
YLPQ motif. Although IL-9Rα expression is not induced by Tax,
expression of IL-9 is activated by Tax via an NF-κB motif in its
proximal promoter.
The IL-9R and common γ chains associated with JAK1
and JAK3, trigger STATs-1, -3 and -5. In addition, they activate
the IRS and RAS-MAPK pathways, although, the physiological
requirement for these pathways in primary cells has not previously
been well documented (17). A
single tyrosine residue (Tyr407) in the IL-9Rα is phosphorylated
following ligand binding to the receptor and activation of
associated JAK1. The mutation of this residue demonstrates that it
is required for IL-9-dependent responses.
As predicted from its initial identification as a
T-cell growth factor, IL-9R is expressed in T-cell lines and
effector T cells, but not in naïve T cells (18,19).
Among the Th-cell subsets, IL-9R exhibits its highest expression in
Th2 and Th17 cells (13). IL-9R is
found on MCs and polymorphonuclear leukocytes (20,21).
Additionally, IL-9R is expressed in non-hematopoietic cells. Since
γc is unlikely to be expressed in these cells, the exact
composition of the receptor in non-hematopoietic cells has not been
clearly defined.
Effect of IL-9 on B cells
Traditionally, IL-9 is described as a T-cell-derived
cytokine with pleiotropic activities on various cell types. More
recently, IL-9 expression has been linked to B cells (22,23).
IL-9 not only exerts effects on B-cell development but also on
function. Transgenic expression of IL-9 recovers the B1 cell
numbers, but not natural IgM production, and results in an increase
of peritoneal CD11b+ B1 cells, in xid mice
(24,25). IL-9 enhances IL-4-mediated IgE and
IgG production from human B cells, but has no effect on IgM
production (26,27). IL-9 exerts similar effects on
germinal center B cells and IL-9Rα expression is greater on such B
cells than on other types of B cells (28).
IL-9-dependent regulation of
hematological malignancies
IL-9 is a T-cell-derived lymphokine that induces the
proliferation of various lymphoid and hemopoietic cells (29). The HTLV-I protein, Tax, is important
in the early stages of adult T-cell leukemia/lymphoma (ATL) based
on altered gene expression, including that of cytokines and their
receptors. Supporting a role for IL-9/IL-9Rα in ATL, a previous
study showed that a neutralizing monoclonal antibody inhibited the
ex vivo spontaneous proliferation of primary ATL cells
obtained from several patients, directed toward IL-9Rα. Freshly
isolated peripheral blood mononuclear cells from these patients
revealed high expression levels of IL-9Rα on their CD14-expressing
monocytes by fluorescence-activated cell sorting analysis.
Furthermore, purified T cells or monocytes did not independently
proliferate from these patients ex vivo, whereas mixtures of
these cell types manifested significant proliferation in a
contact-dependent manner. Overall, these results suggested that
primary ATL cells support the action of IL-9Rα/CD14-expressing
monocytes via IL-9, which subsequently supports the ex vivo
spontaneous proliferation of malignant T cells. In conclusion,
these results supported the theory that IL-9 and its receptor are
involved in ATL through a paracrine mechanism (17,30).
Previous studies analyzing the culture supernatants
of peripheral blood mononucleated cells (PBMCs) from the early
phases of ATL patients with spontaneous proliferation using the
cytokine-dependent indicator cell line, NK-92, have revealed that
the majority of six-day culture supernatants of PBMCs from ATL
patients contain high amounts of IL-9 (31). The presence of IL-9 in the culture
supernatants was also confirmed by ELISA analysis. Furthermore, in
certain ATL patients within this group, the spontaneous
proliferation was blocked by a monoclonal antibody against IL-9Rα.
This suggested that the IL-9/IL-9R system may be involved in the
expansion of the HTLV-I-infected CD4+ T cells of
specific patients in the early stages of ATL (32). IL-9 is a Th2 cytokine and IL-9 is
produced in >80% of smoldering/chronic ATL ex vivo PBMC
cultures, which has previously shown that HTLV-1 Tax transactivates
IL-9 expression in HTLV-1-infected T-cell lines (30). Prevention of histone acetylation by
the histone acetyltransferase inhibitor, curcumin, diminished PU.1
expression following IL-9-inducing stimulation. Autocrine/paracrine
cytokine stimulation of leukemic cell proliferation has been
identified in patients with smoldering/chronic ATL that may be
targeted for treatment. The etiologic agent of ATL is HTLV-I.
Anaplastic lymphoma kinase (ALK) has been implicated
in the growth of neoplastic cells in malignant lymphomas. Although
IL-9 has been implicated in the growth of normal MCs, little is
known concerning pro-oncogenic molecules and conditions triggering
differentiation and growth of MC to lead to the histopathological
image of overt mastocytosis. Certain previous studies have
described that transplantation of nucleophosmin
(NPM)-ALK-transplanted mouse bone marrow progenitors into
lethally irradiated IL-9 transgenic mice not only results in
lymphoma formation, but also in the development of a neoplastic
disease exhibiting histopathological features of systemic
mastocytosis. These include multifocal dense MC infiltrates,
occasionally with devastating growth in visceral organs.
Transplantation of NPM-ALK-transduced progenitors into
normal mice or maintenance of IL-9 transgenic mice without NPM-ALK
both resulted in MC hyperplasia, but not in mastocytosis.
Neoplastic MCs in mice are not only exhibited in IL-9, but also the
IL-9R, as was found in human neoplastic MCs. Overall, the data show
that neoplastic MCs express IL-9Rs. In addition, IL-9 and NPM-ALK
upregulate MC production in vivo and the two ‘hits’ act in
concert to induce a mastocytosis-like disease in mice (33). These results may have pathogenetic
and clinical implications, and are consistent with the observation
that neoplastic MCs in advanced systemic mastocytosis markedly
express NPM and multiple ‘lymphoid’ antigens, including CD25 and
CD30.
3. Characteristics and expression of
STATs
STAT3 target genes
STAT3 is a cytoplasmic transcription factor and a
member of the STAT family. Aberrant STAT3 activation is considered
a molecular abnormality that supports the tumor phenotype and is
detected with high frequency in hematological malignancies. STAT3
is involved in embryonic stem (ES) cell self-renewal (stemness) of
certain mammalian cell types and species. Generally, STAT3-mediated
transcription directs cells into cell survival and cell cycle
progression. STAT3 is involved in cellular transformation and
tumorigenesis (34). However, the
molecular mechanisms leading to the aberrant STAT3 activation and
STAT3-mediated transformation and tumorigenesis remain unclearly
defined.
STAT3 proteins are also activated via cytoplasmic
kinases of the Src kinase family and via the tyrosine kinase
activity of various growth factor receptors. Cytokines activate
STAT3 proteins differentially, utilizing the gp130 receptor
component, including ciliary neurotrophic factor, oncostatin M,
leukemia-inhibitory factor (LIF) and IL-6. STAT3 proteins are also
activated by growth hormones, including thrombopoietin, granulocyte
colony-stimulating factor, granulocyte-macrophage
colony-stimulating factor, basic fibroblast growth factor and a
number of ILs (35). The results of
previous proteomic studies have uncovered an interdependence of
STAT3 signaling and members of the Rho family of small GTPases,
including Rac1, Cdc42 and RhoA. Specifically, Rac1, acting in
complex with the male germ cell, RacGAP, promotes tyrosine
phosphorylation of STAT3 by the IL6-receptor family/JAK complex, as
well as its translocation to the nucleus. Evidence has further
demonstrated that the mutational activation of Rac1 and Cdc42
results in STAT3 activation, which occurs, in part, through the
upregulation of the IL6 family of cytokines that, in turn,
stimulate STAT3 through JAKs. Notably, previous studies have also
shown that the engagement of cadherins and cell-to-cell adhesion
molecules, specifically, induce a marked increase in Rac1 and Cdc42
protein levels and activity, which, in turn, results in STAT3
activation (36).
STAT3 is tightly integrated into the gene regulatory
mechanism of pluripotency. Previously, Kidder et al
(37) applied chromatin
immunoprecipitation (ChIP) sequencing technology to use promoter
arrays of mouse ES cells (mESCs) that cover 28,000 promoter regions
in the genome. The authors found 948 putative target genes for
STAT3, with 29 of these genes co-occupied by Oct4 and
Nanog. In addition, Chen et al (38) used the ChIP-chip approach to map the
binding sites for STAT3 in mESCs. The authors identified 2,546
genomic sites where STAT3 was bound and approximately one-third
(718) of these loci were bound by Oct4, Sox2 and Nanog.
The target gene list contains transcriptionally
active and inactive genes. A number of pluripotency genes, such as
Oct4 and Nanog, exist among the STAT3-binding sites
(39). The inactive genes include
developmentally regulated tissue-specific genes, including
Gata3 (ectoderm-specific), Foxa2, Gata4 (the
two endoderms), T (brachyury), LIM homeobox protein 1
(mesoderm) and Eomes (trophectoderm). Although the present
review did not identify whether STAT3 suppresses these genes, it is
possible that STAT3 mediates the suppression of differentiation
genes and that this mechanism may be one method in which LIF
prevents the differentiation of mESCs into endoderm and mesoderm
lineages (40). In a previous
study, although the authors did not report whether any of these 113
suppressed genes included the aforementioned developmentally
regulated genes, it was found that STAT3 was bound to 113 genes
that were also bound by subunits of the polycomb repressive complex
2, such as Suz12 and Eed (37). In
general, STAT proteins function as transcriptional activators, but
STAT1 is known to suppress the transcription of matrix
metalloproteinases and cell-cycle genes (c-Myc, cyclin D and cyclin
A) (41). Therefore, STAT3 may also
function as a suppressor. Along an associated line of inquiry,
Bourillot et al (42)
knocked down 22 STAT3 target genes and found that 16 induced
activation of endodermal genes and one activated mesodermal genes.
These observations are consistent with the theory that STAT3
contributes to the prevention of mESC differentiation by
suppressing lineage-specific genes.
STAT3-binding proteins and gene
activation
An additional method to understanding how STAT3
regulates gene expression is to identify proteins that interact
with STAT3. The interacting proteins include the transcription
factors, NF-κB (43) and c-Jun
(44), the coactivators, nuclear
receptor coactivator 1/SRC1a (45)
and Ctr9 (46), and the chromatin
remodeling adenylpyrophosphatase (ATPase) brahma-related gene 1
(Brg1) (47,48), p300/CBP. These interactions have
been previously studied outside the field of stem cell biology, but
similar interactions are likely to occur in mESCs since specific
interacting partners are colocalized with STAT3 on the mESC genome.
The transactivation domain of STAT3 at the C-terminus interacts
with a number of chromatin proteins, including p300/CBP and is
colocalized with STAT3 on a number of pluripotency genes (38).
Of particular interest is Brg1, a catalytic subunit
of the switch/sucrose non-fermentable ATPase complex that is
associated with pluripotency at several levels. Brg1 is involved in
chromatin relaxation (99); however, the role of Brg1 in mESCs is
not limited to chromatin relaxation. Previously, it was
demonstrated that the ESC-specific chromatin remodeling esBAF
complex contains Brg1 (50,51). esBAF is colocalized with STAT3
throughout the genome, including in pluripotency genes. Although
esBAF binds to a number of pluripotency genes in ESCs, it maintains
pluripotency primarily by suppressing differentiation-specific
genes (49). Additionally, Brg1
facilitates the binding of Oct4 to its target genes and
increases the efficiency of the dedifferentiation of fibroblasts to
a pluripotent state (52). Although
little is known concerning the details of the interaction between
Brg1 and STAT3 during LIF stimulation, recruitment of Brg1 occurs
prior to or following STAT3 binding to its target genes, depending
on the particular IL-6 target genes. For specific IL-6 target
genes, Brg1 is constitutively bound and its presence is necessary
for the recruitment of STAT3 (47).
For other IL-6 target genes, STAT3 binds first to its DNA and then
Brg1 is recruited depending on the presence of STAT3 (48).
Ctr9 is a subunit of the Paf1
complex
The STAT3-Ctr9 interaction is highly significant for
pluripotency of mESCs since Ctr9 indirectly induces multiple
histone modifications, including trimethylation of Lys4 and Lys36,
dimethylation of Lys79 on histone H3 and ubiquitination of histone
H2B, all of which are important for gene activation (53). Consistent with this, the level of
trimethylation of Lys4 on histone H3 on specific IL-6-inducible
genes is dependent on the presence of Ctr9 (46). It appears that the interaction
between STAT3 and Ctr9 is important for the recruitment of STAT3 to
its target genes in this case. Although it is not known whether LIF
also induces the interaction between STAT3 and Ctr9 in mESCs,
confirmation is likely to provide the first molecular link between
LIF and epigenetic modifications in ESCs. The Paf1 complex also
binds to Oct4 in mESCs (54,55),
but it is unknown whether STAT3 is relevant to this binding.
STAT6 target genes
STAT6 is a transcription factor and mainly
responsible for their own transcriptional effects. It is primarily
activated by IL-4 and IL-13 and its C-terminal Src homology 2 (SH2)
domain is a specific phosphorylated receptor site. STAT6 is
phosphorylated on Tyr641 and is subsequently activated. The STAT6
protein then dimerizes and translocates to the nucleus where it
binds to STAT regulatory elements and regulates transcription in
association with other transcription factors in its activated form
(56). Previously, STAT6 has been
reported to be constitutively activated in HL-derived cell lines
(57).
Previously performed high-throughput sequencing of
chromatin immunoprecipitated DNA has identified genes bound by
STAT6. In addition, a previous study compared genes bound by STAT6
in wild-type and STAT6−/− Th2 cells and these results
were compared with epigenetic modifications across the genome
(58). In the current study,
H3K4me3 was colocalized with 60% of the binding sites for STAT6.
Various permissive epigenetic marks were coincided with specific
STAT6-bound regions and IL-4, Gata3, IL-24, phospholipase C δ 1 and
homeodomain interacting protein kinase 2 were included in the
corresponding genes (58). In an
additional study, human Th2 cells were used and compared with the
STAT6 binding to genes between cells where the expression of STAT6
was knocked down by RNA interference and cells with normal STAT6
expression (59). In the present
study, a kinetic analysis was performed and the identity of
STAT6-dependent genes during the Th2 polarization process was
determined. It was found that 80% of IL-4 regulated genes were
dependent on STAT6 at the 48-h time point. Specific genes regulated
by STAT6 included Gata3, CRTH2, IL-24, lymphotoxin β, suppressor of
cytokine signaling (SOCS) 1. A resource which may be used for
future studies to define further roles of STAT6 in T and B cells is
provided with high-throughput screening for STAT6-regulated genes.
Emerging evidence shows that STAT6 functions in other immune cells,
as well as other non-immune cells. STAT6 is likely to be
significant in determining the nature of genes that are regulated
by STAT6 in these tissues.
STAT6-binding proteins and gene
activation
STAT6 functions may be analyzed using mice with
disrupted Stat6 alleles. A previous mouse study demonstrated
that STAT6 is critical for a number of responses in T cells,
including the development of Th2 cells and IL-4-stimulated
proliferative responses. The expression of Th2 cytokines, including
IL-4, -5 and -13, was diminished in Stat6−/− mice
(60). The expression of Gata3, the
master regulator of Th2 differentiation, may be regulated by STAT6
(61). STAT6 is also required for
the development of IL-9-secreting T cells (62–64).
The mechanisms by which STAT6 regulates T-cell proliferation
include decreasing the expression of p27Kip1, a known
cyclin-dependent kinase inhibitor, which may be at the
transcriptional and post-translational levels (65,66).
STAT6 is required for cytotoxic T2-cell differentiation, as the
production of IL-4 and IL-5 is completely lost with STAT6
deficiency in CD8 cells (67).
Overall, STAT6 is required for IL-4-stimulated T-cell
functions.
In B cells, STAT6 promotes immunoglobulin class
switching to IgE and IgG1, as well as promoting the expression of
specific cell surface molecules responsible for antigen
presentation by B cells (68). In a
previous study, the levels of IgE were evidently reduced in
STAT6-deficient mice when the mice were sensitized with antigen or
infected with N. brasiliensis (61). No differences were identified in
immunoglobulin class switching to IgG1 when
STAT6−/− and control mice were immunized with
IgD, but the levels of IgG1 were reduced in STAT6-deficient mice,
an infection model with N. brasiliensis or S. mansoni
(69). The expression of several
cell surface molecules responsible for antigen presentation by B
cells is induced by IL-4, including MHC II, CD80 and CD86. Previous
studies using STAT6−/− B cells and mice
expressing a constitutive form of STAT6 showed that the
IL-4-mediated induction of these molecules is dependent on STAT6
(60,68). The expression of other cell surface
molecules, such as CD23 and IL-4R-α, are also induced by STAT6
(68). CD23 is the low-affinity Fc
receptor for IgE and is also a B-cell differentiation marker. The
induction of IL-4R-α by STAT6 indicates that STAT6 promotes an
autocrine-positive feedback loop for IL-4-dependent signaling. In B
cells, STAT6 is required for IL-4-stimulated proliferation, similar
to the previously described role of T cells (61). In addition, apoptosis is prevented
in B cells by IL-4 in a STAT6-dependent manner (70).
STAT6 also functions in macrophages and dendritic
cells, in addition to a requirement in T and B cells. STAT6
mediates IL-13-induced expression of genes, including MHC class II,
and promotes IL-4-induced differentiation of alternatively
activated macrophages (AAM) in macrophages (71,72).
STAT6 activity in AAMs is associated with the suppression of T-cell
proliferation (73). Currently, one
previous study has shown that STAT6 facilitates the transcription
mediated by the peroxisome proliferator-activated-γ receptor in
macrophages and dendritic cells (74). STAT6 is capable of downregulating
the production of IL-10 and promoting the production of IL-12 to
promote a Th1 response in dendritic cells (75). Thus, STAT6 is vital in regulating
the balance of inflammatory and allergic immune responses.
STAT pathway in hematological
malignancies
STAT proteins are activated by a wide range of
cytokines and growth factors, typically via cytokines through the
JAK family of tyrosine kinases (76).
Aprevious study revealed a close cross-talk
correlation between STAT3 and phosphoinositide 3-kinase (PI3K)
(77). STAT3 in PI3K-transformed
murine cells is phosphorylated on Y705 and activated in a
PI3K-dependent manner. Dominant-negative STAT3 interferes with
PI3K-induced oncogenic transformation. In PI3K-transformed murine
cells, phosphorylation of STAT3 is mediated by the TEC kinase, BMX.
STAT3 is important in PI3K-driven oncogenic transformation and
marks BMX as a promising therapeutic target that may enhance the
efficacy of PI3K inhibitors. The PI3K-mammalian target of rapamycin
and STAT3 signaling pathways represent two different regulatory
networks. The functional link between these pathways is significant
for the understanding of the PI3K- and STAT3-driven oncogenic
mechanisms and identifies the TEC kinase, BMX, as a new cancer
target.
STAT3 mutations unify the pathogenesis of chronic
lymphoproliferative disorders of natural killer (NK) cells and
T-cell large granular lymphocyte leukemia. STAT3 gene mutations are
present in T and NK cell diseases. Mutations have been found in
exons 21 and 20, encoding the SH2 domain. Constitutive STAT3,
Tyr705 and Ser727 phosphorylation caused by the autocrine secretion
of IL-6 exist in acute myeloid leukemia cells, murine plasmacytomas
and hybridomas. In addition, STAT3-mediated constitutive expression
of SOCS-3 is present in cutaneous T-cell lymphoma (78).
STAT3 is crucial in promoting the progression of
hematological malignancies, including chronic lymphocytic leukemia
(CLL). In CLL, STAT3 is constitutively phosphorylated on serine
727, regardless of blood count, disease stage or treatment status
and not on the tyrosine 705 residue; however, the biological
significance of serine-phosphorylated STAT3 (pSTAT3) is not known.
A previous study demonstrated that constitutive serine pSTAT3
translocates to the nucleus by the karyopherin-β nucleocytoplasmic
system and binds to DNA. Dephosphorylation of inducible tyrosine
pSTAT3 did not affect STAT3-DNA binding. Furthermore, infection of
CLL cells with lentiviral STAT3-small hairpin RNA (shRNA) reduced
the expression of several STAT3-regulated survival and
proliferation genes and induced apoptosis. Overall, the results
suggested that constitutive phosphorylation of STAT3 on the serine
727 residue is a hallmark of CLL and that STAT3 may be considered a
therapeutic target in this disease (79).
Targeting sphingosine-1-phosphate
(S1P)/sphingosine-1-phosphate receptor 1 (S1PR1) using a clinically
relevant and available drug or other approaches is potentially an
effective, new therapeutic modality for treating the activated B
cell-like subtype of diffuse large B-cell lymphoma, a subset of
lymphoma that is less responsive to current available therapies.
Studies have shown that using S1PR1 shRNA, a G protein-coupled
receptor for S1P or FTY720, an antagonist of S1P that is used
clinically for other indications, inhibits S1PR1 expression and
downregulates STAT3 activity, causing growth inhibition of the
B-cell lymphoma tumor cells in vitro and in vivo
(80).
Recurrent mutations of the STAT6 DNA binding domain
strongly support the involvement of STAT6 in the pathogenesis of
this aggressive B-cell lymphoma (81). The STAT6 signaling pathway,
activated by the cytokines IL-4 and IL-13, induces expression of
the Epstein-Barr virus (EBV)-encoded protein LMP-1 in absence of
the EBV nuclear antigen 2; implicating the type II EBV latent gene
expression in HL (82).
4. Role of STAT3 and STAT6 in IL-9
production and its immune function
STAT3 is a central cytoplasmic transcription factor
that is activated by the phosphorylation of a conserved tyrosine
residue in response to oncogenic proteins and extracellular
signals, such as cytokines and growth factors (83). STAT3 regulates a number of genes
that are critical to tumor cell survival and proliferation,
angiogenesis, invasion, metastasis and immune evasion (84,85).
Previous extensive studies have demonstrated that inappropriate
activation of STAT3 occurs at a high frequency in a wide variety of
human cancers, including leukemia and lymphoma (86–89).
Co-expression of the IL-9Rα chain promotes JAK1 mutant
phosphorylation and STATs activation, including STAT1, STAT3 and
STAT5.
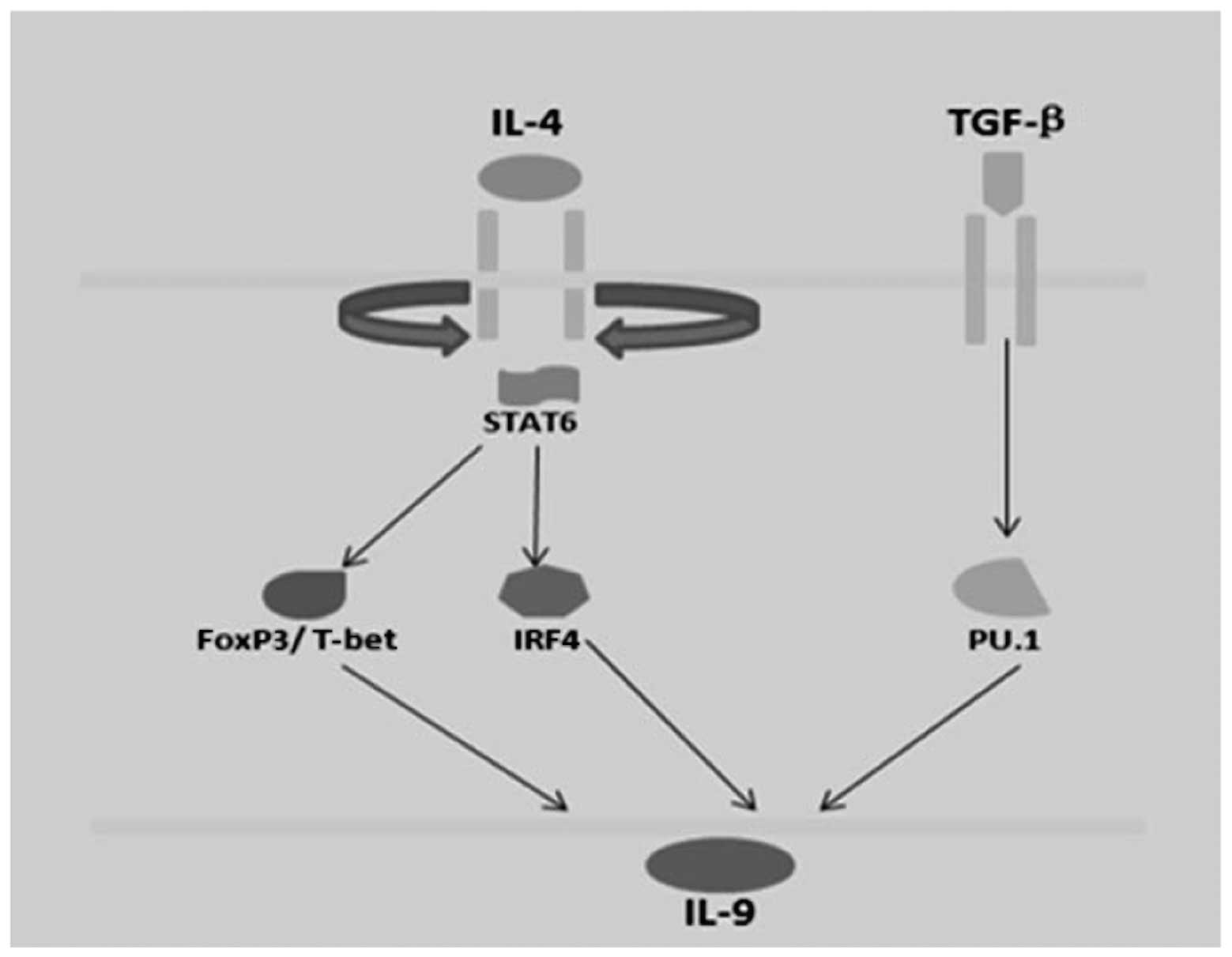
In addition, Th2 and Th9 cells are described as
IL-9-secreting cells, and culture with TGF-β1 may increase IL-9
production. Th9 cells are derived in culture with a combination of
TGF-β1 and IL-4. In addition, Th9 cells are associated with Th2
cells in that they require STAT6 and Gata3 for development, but
exhibit reduced expression of Th2 cytokines. STAT6 is required for
the expression of Gata3 in Th9 cells (90). Ectopic expression of Gata3 reduced
IL-9 production in wild-type cells but did not induce IL-9
production when transduced into STAT6−/− Th9
cultures, suggesting that it is not directly regulating the
Il9 gene. It is possible that Gata3 is an intermediate in
the STAT6-dependent reduction. IL-9 is not expressed by Th2 and Th9
cells in the absence of STAT6 expression (Fig. 2).
5. Conclusions and future directions
IL-9 is a multifunctional cytokine secreted by Th2
lymphocytes. The IL-9R and common γ chains associated with JAK1 and
JAK3 trigger STAT-1, -3 and -5. In addition, dysregulated IL-9
response leads to autonomous cell growth and malignant
transformation of lymphoid cells associated with constitutive
activation of the JAK/STAT pathway in vitro. The most recent
example of a Th subset that requires multiple balanced signals to
develop is Th9 cells that secrete IL-9. Th9 cells develop following
exposure to TGFβ and IL-4, whereas TGFβ alone promotes the
differentiation of Treg cells and IL-4 stimulates Th2 development
(91). The integration of the two
signals result in a Th subset that exhibits lower Foxp3 expression
than Treg cultures and lower Th2 cytokine production than Th2
cells, but increased production of IL-9. However, it remains
unclear how the integration of each signal results in the unique
Th9 phenotype. In any case, the role of IL-9 and STATs has
important implications for pathophysiology and treatment in
hematological malignancies.
Acknowledgements
The present study was partly supported by grants
from the Natural Science Foundations of Shandong Province, China
(nos. Y2007C053 and ZR2009CM059), the Technology Development
Projects of Shandong Province, China (nos. 2007GG10002008,
2008GG2NS02018 and 2010GSF10250), the Program for Outstanding
Medical Academic Leader of Shandong Province, China and the Taishan
Scholar Foundation of Shandong Province, China.
References
|
1
|
Namkung JH, Lee JE, Kim E, et al: An
association between IL-9 and IL-9 receptor gene polymorphisms and
atopic dermatitis in a Korean population. J Dermatol Sci. 62:16–21.
2011.PubMed/NCBI
|
|
2
|
Knoops L and Renauld JC: IL-9 and its
receptor: from signal transduction to tumorigenesis. Growth
Factors. 22:207–215. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Putheti P, Awasthi A, Popoola J, Gao W and
Strom TB: Human CD4 memory T cells can become CD4+IL-9+ T cells.
PLoS One. 5:e87062010.
|
|
4
|
van den Ham HJ, de Waal L, Andeweg AC and
de Boer RJ: Identification of helper T cell master regulator
candidates using the polar score method. J Immunol Methods.
361:98–109. 2010.
|
|
5
|
Chang HC, Han L, Jabeen R, Carotta S, Nutt
SL and Kaplan MH: PU.1 regulates TCR expression by modulating
GATA-3 activity. J Immunol. 183:4887–4894. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chang HC, Zhang S, Thieu VT, Slee RB,
Bruns HA, Laribee RN, Klemsz MJ and Kaplan MH: PU.1 expression
delineates heterogeneity in primary Th2 cells. Immunity.
22:693–703. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang HC, Sehra S, Goswami R, Yao W, Yu Q,
Stritesky GL, Jabeen R, McKinley C, Ahyi AN, Han L, et al: The
transcription factor PU.1 is required for the development of
IL-9-producing T cells and allergic inflammation. Nat Immunol.
11:527–534. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Staudt V, Bothur E, Klein M, Lingnau K,
Reuter S, Grebe N, Gerlitzki B, Hoffmann M, Ulges A, Taube C, et
al: Interferon-regulatory factor 4 is essential for the
developmental program of T helper 9 cells. Immunity. 33:192–202.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ahyi AN, Chang HC, Dent AL, Nutt SL and
Kaplan MH: IFN regulatory factor 4 regulates the expression of a
subset of Th2 cytokines. J Immunol. 183:1598–1606. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brustle A, Heink S, Huber M, Rosenplanter
C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T and Lohoff M:
The development of inflammatory T(H)-17 cells requires
interferon-regulatory factor 4. Nat Immunol. 8:958–966. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lohoff M, Mittrucker HW, Prechtl S,
Bischof S, Sommer F, Kock S, Ferrick DA, Duncan GS, Gessner A and
Mak TW: Dysregulated T helper cell differentiation in the absence
of interferon regulatory factor 4. Proc Natl Acad Sci USA.
99:11808–11812. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hültner L, Kölsch S, Stassen M, Kaspers U,
Kremer JP, Mailhammer R, Moeller J, Broszeit H and Schmitt E: In
activated mast cells, IL-1 up-regulates the production of several
Th2-related cytokines including IL-9. J Immunol. 164:5556–5563.
2000.PubMed/NCBI
|
|
13
|
Stassen M, Arnold M, Hültner L, Müller C,
Neudörfl C, Reineke T and Schmitt E: Murine bone marrow-derived
mast cells as potent producers of IL-9: costimulatory function of
IL-10 and kit ligand in the presence of IL-1. J Immunol.
164:5549–5555. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stassen M, Müller C, Arnold M, Hültner L,
Klein-Hessling S, Neudörfl C, Reineke T, Serfling E and Schmitt E:
IL-9 and IL-13 production by activated mast cells is strongly
enhanced in the presence of lipopolysaccharide: NF-kappa B is
decisively involved in the expression of IL-9. J Immunol.
166:4391–4398. 2001. View Article : Google Scholar
|
|
15
|
Stassen M, Klein M, Becker M, Bopp T,
Neudörfl C, Richter C, Heib V, Klein-Hessling S, Serfling E, Schild
H and Schmitt E: p38 MAP kinase drives the expression of mast
cell-derived IL-9 via activation of the transcription factor
GATA-1. Mol Immunol. 44:926–933. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Osterfeld H, Ahrens R, Strait R, Finkelman
FD, Renauld JC and Hogan SP: Differential roles for the IL-9/IL-9
receptor alpha-chain pathway in systemic and oral antigen-induced
anaphylaxis. J Allergy Clin Immunol. 125:469–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Demoulin JB, Louahed J, Dumoutier L,
Stevens M and Renauld JC: MAP kinase activation by interleukin-9 in
lymphoid and mast cell lines. Oncogene. 22:1763–1770. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cosmi L, Liotta F, Angeli R, Mazzinghi B,
Santarlasci V, Manetti R, Lasagni L, Vanini V, Romagnani P, Maggi
E, et al: Th2 cells are less susceptible than Th1 cells to the
suppressive activity of CD25+ regulatory thymocytes because of
their responsiveness to different cytokines. Blood. 103:3117–3121.
2004.
|
|
19
|
Druez C, Coulie P, Uyttenhove C and Van
Snick J: Functional and biochemical characterization of mouse
P40/IL-9 receptors. J Immunol. 145:2494–2499. 1990.PubMed/NCBI
|
|
20
|
Abdelilah S, Latifa K, Esra N, Cameron L,
Bouchaib L, Nicolaides N, Levitt R and Hamid Q: Functional
expression of IL-9 receptor by human neutrophils from asthmatic
donors: role in IL-8 release. J Immunol. 166:2768–2774. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kearley J, Erjefalt JS, Andersson C,
Benjamin E, Jones CP, Robichaud A, Pegorier S, Brewah Y, Burwell
TJ, Bjermer L, et al: IL-9 governs allergen-induced mast cell
numbers in the lung and chronic remodeling of the airways. Am J
Respir Crit Care Med. 183:865–875. 2011. View Article : Google Scholar
|
|
22
|
Nowak EC, Weaver CT, Turner H, Begum-Haque
S, Becher B, Schreiner B, Coyle AJ, Kasper LH and Noelle RJ: IL-9
as a mediator of Th17-driven inflammatory disease. J Exp Med.
206:1653–1660. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu LF, Lind EF, Gondek DC, Bennett KA,
Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J,
et al: Mast cells are essential intermediaries in regulatory T-cell
tolerance. Nature. 442:997–1002. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Knoops L, Louahed J and Renauld JC:
IL-9-induced expansion of B-1b cells restores numbers but not
function of B-1 lymphocytes in xid mice. J Immunol. 172:6101–6106.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vink A, Warnier G, Brombacher F and
Renauld JC: Interleukin 9-induced in vivo expansion of the B-1
lymphocyte population. J Exp Med. 189:1413–1423. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dugas B, Renauld JC, Pene J, Bonnefoy JY,
Peti-Frère C, Braquet P, Bousquet J, Van Snick J and Mencia-Huerta
JM: Interleukin-9 potentiates the interleukin-4-induced
immunoglobulin (IgG, IgM and IgE) production by normal human B
lymphocytes. Eur J Immunol. 23:1687–1692. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petit-Frere C, Dugas B, Braquet P and
Mencia-Huerta JM: Interleukin-9 potentiates the
interleukin-4-induced IgE and IgG1 release from murine B
lymphocytes. Immunology. 79:146–151. 1993.PubMed/NCBI
|
|
28
|
Fawaz LM, Sharif-Askari E, Hajoui O,
Soussi-Gounni A, Hamid Q and Mazer BD: Expression of IL-9 receptor
alpha chain on human germinal center B cells modulates IgE
secretion. J Allergy Clin Immunol. 120:1208–1215. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Renauld JC, Druez C, Kermouni A, Houssiau
F, Uyttenhove C, Van Roost E and Van Snick J: Expression cloning of
the murine and human interleukin 9 receptor cDNAs. Proc Natl Acad
Sci USA. 89:5690–5694. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Petrus M, Bryant BR, Phuc Nguyen
V, Stamer M, Goldman CK, Bamford R, Morris JC, Janik JE and
Waldmann TA: Induction of the IL-9 gene by HTLV-I Tax stimulates
the spontaneous proliferation of primary adult T-cell leukemia
cells by a paracrine mechanism. Blood. 111:5163–5172. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Umezu-Goto M, Kajiyama Y, Kobayashi N,
Kaminuma O, Suko M and Mori A: IL-9 production by peripheral blood
mononuclear cells of atopic asthmatics. Int Arch Allergy Immunol.
143(Suppl 1): 76–79. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen J, Petrus M, Bryant BR, Nguyen VP,
Goldman CK, Bamford R, Morris JC, Janik JE and Waldmann TA:
Autocrine/paracrine cytokine stimulation of leukemic cell
proliferation in smoldering and chronic adult T-cell leukemia.
Blood. 116:5948–5956. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Merz H, Kaehler C, Hoefig KP, et al:
Interleukin-9 (IL-9) and NPM-ALK each generate mast cell
hyperplasia as single ‘hit’ and cooperate in producing a
mastocytosis-like disease in mice. Oncotarget. 1:104–119.
2010.PubMed/NCBI
|
|
34
|
Lin Q, Lai R, Chirieac LR, Li C, Thomazy
VA, Grammatikakis I, Rassidakis GZ, Zhang W, Fujio Y, Kunisada K,
et al: Constitutive activation of JAK3/STAT3 in colon carcinoma
tumors and cell lines: inhibition of JAK3/STAT3 signaling induces
apoptosis and cell cycle arrest of colon carcinoma cells. Am J
Pathol. 167:969–980. 2005. View Article : Google Scholar
|
|
35
|
Dahéron L, Opitz SL, Zaehres H, Lensch MW,
Andrews PW, Itskovitz-Eldor J and Daley GQ: LIF/STAT3 signaling
fails to maintain self-renewal of human embryonic stem cells. Stem
Cells. 22:770–778. 2004.PubMed/NCBI
|
|
36
|
Raptis L, Arulanandam R, Geletu M and
Turkson J: The R(h)oads to Stat3: Stat3 activation by the Rho
GTPases. Exp Cell Res. 317:1787–1795. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kidder BL, Yang J and Palmer S: STAT3 and
c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS
One. 3:e39322008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen X, Xu H, Yuan P, Fang F, Huss M, Vega
VB, Wong E, Orlov YL, Zhang W, Jiang J, et al: Integration of
external signaling pathways with the core transcriptional network
in embryonic stem cells. Cell. 133:1106–1117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kuznetsov VA, Singh O and Jenjaroenpun P:
Statistics of protein-DNA binding and the total number of binding
sites for a transcription factor in the mammalian genome. BMC
Genomics. 11(Suppl 1): S122010. View Article : Google Scholar
|
|
40
|
Ying QL, Nichols J, Chambers I and Smith
A: BMP induction of Id proteins suppresses differentiation and
sustains embryonic stem cell self-renewal in collaboration with
STAT3. Cell. 115:281–292. 2003. View Article : Google Scholar
|
|
41
|
Ramana CV, Chatterjee-Kishore M, Nguyen H
and Stark GR: Complex roles of STAT1 in regulating gene expression.
Oncogene. 19:2619–2627. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bourillot PY, Aksoy I, Schreiber V, Wianny
F, Schulz H, Hummel O, Hubner N and Savatier P: Novel STAT3 target
genes exert distinct roles in the inhibition of mesoderm and
endoderm differentiation in cooperation with Nanog. Stem Cells.
27:1760–1771. 2009. View Article : Google Scholar
|
|
43
|
Yu Z, Zhang W and Kone BC: Signal
transducers and activators of transcription 3 (STAT3) inhibits
transcription of the inducible nitric oxide synthase gene by
interacting with nuclear factor κB. Biochem J. 367:97–105.
2002.PubMed/NCBI
|
|
44
|
Zhang X, Wrzeszczynska MH, Horvath CM and
Darnell JE Jr: Interacting regions in STAT3 and c-Jun that
participate in cooperative transcriptional activation. Mol Cell
Biol. 19:7138–7146. 1999.
|
|
45
|
Giraud S, Bienvenu F, Avril S, Gascan H,
Heery DM and Coqueret O: Functional interaction of STAT3
transcription factor with the coactivator NcoA/SRC1a. J Biol Chem.
277:8004–8011. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Youn MY, Yoo HS, Kim MJ, Hwang SY, Choi Y,
Desiderio SV and Yoo JY: hCTR9, a component of Paf1 complex,
participates in the transcription of interleukin 6-responsive genes
through regulation of STAT3-DNA interactions. J Biol Chem.
282:34727–34734. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ni Z and Bremner R: Brahma-related gene
1-dependent STAT3 recruitment at IL-6-inducible genes. J Immunol.
178:345–351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Giraud S, Hurlstone A, Avril S and
Coqueret O: Implication of BRG1 and cdk9 in the STAT3-mediated
activation of the p21waf1 gene. Oncogene. 23:7391–7398. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ho L, Jothi R, Ronan JL, Cui K, Zhao K and
Crabtree GR: An embryonic stem cell chromatin remodeling complex,
esBAF, is an essential component of the core pluripotency
transcriptional network. Proc Natl Acad Sci USA. 106:5187–5191.
2009. View Article : Google Scholar
|
|
50
|
Ho L, Ronan JL, Wu J, Staahl BT, Chen L,
Kuo A, Lessard J, Nesvizhskii AI, Ranish J and Crabtree GR: An
embryonic stem cell chromatin remodeling complex, esBAF, is
essential for embryonic stem cell self-renewal and pluripotency.
Proc Natl Acad Sci USA. 106:5181–5186. 2009. View Article : Google Scholar
|
|
51
|
Singhal N, Graumann J, Wu G, Araúzo-Bravo
MJ, Han DW, Greber B, Gentile L, Mann M and Schöler HR:
Chromatin-remodeling components of the BAF complex facilitate
reprogramming. Cell. 141:943–955. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guiter C, Dusanter-Fourt I, Copie-Bergman
C, Boulland ML, Le Gouvello S, Gaulard P, Leroy K and Castellano F:
Constitutive STAT6 activation in primary mediastinal large B-cell
lymphoma. Blood. 104:543–549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gerber M and Shilatifard A:
Transcriptional elongation by RNA polymerase II and histone
methylation. J Biol Chem. 278:26303–26306. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ding L, Paszkowski-Rogacz M, Nitzsche A,
Slabicki MM, Heninger AK, de Vries I, Kittler R, Junqueira M,
Shevchenko A, Schulz H, et al: A genome-scale RNAi screen for Oct4
modulators defines a role of the Paf1 complex for embryonic stem
cell identity. Cell Stem Cell. 4:403–415. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ponnusamy MP, Deb S, Dey P, Chakraborty S,
Rachagani S, Senapati S and Batra SK: RNA polymerase II associated
factor 1/PD2 maintains self-renewal by its interaction with Oct3/4
in mouse embryonic stem cells. Stem Cells. 27:3001–3011.
2009.PubMed/NCBI
|
|
56
|
Lessard JA and Crabtree GR: Chromatin
regulatory mechanisms in pluripotency. Annu Rev Cell Dev Biol.
6:503–532. 2010. View Article : Google Scholar
|
|
57
|
Skinnider BF, Elia AJ, Gascoyne RD,
Patterson B, Trumper L, Kapp U and Mak TW: Signal transducer and
activator of transcription 6 is frequently activated in Hodgkin and
Reed-Sternberg cells of Hodgkin lymphoma. Blood. 99:618–626. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wei L, Vahedi G, Sun HW, Watford WT,
Takatori H, Ramos HL, Takahashi H, Liang J, Gutierrez-Cruz G, Zang
C, et al: Discrete roles of STAT4 and STAT6 transcription factors
in tuning epigenetic modifications and transcription during T
helper cell differentiation. Immunity. 32:840–851. 2010. View Article : Google Scholar
|
|
59
|
Elo LL, Järvenpää H, Tuomela S, Raghav S,
Ahlfors H, Laurila K, Gupta B, Lund RJ, Tahvanainen J, Hawkins RD,
et al: Genome-wide profiling of interleukin-4 and STAT6
transcription factor regulation of human Th2 cell programming.
Immunity. 32:852–862. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Takeda K, Tanaka T, Shi W, Matsumoto M,
Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T and
Akira S: Essential role of STAT6 in IL-4 signalling. Nature.
380:627–630. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ansel KM, Djuretic I, Tanasa B and Rao A:
Regulation of Th2 differentiation and Il4 locus accessibility. Annu
Rev Immunol. 24:607–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dardalhon V, Awasthi A, Kwon H, Galileos
G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC,
Khoury S, Oukka M and Kuchroo VK: IL-4 inhibits TGF-beta-induced
Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+
Foxp3(−) effector T cells. Nat Immunol. 9:1347–1355.
2008.PubMed/NCBI
|
|
63
|
Perumal NB and Kaplan MH: Regulating Il9
transcription in T helper cells. Trends Immunol. 32:146–150. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Veldhoen M, Uyttenhove C, van Snick J,
Helmby H, Westendorf A, Buer J, Martin B, Wilhelm and Stockinger B:
Transforming growth factor-beta ‘reprograms’ the differentiation of
T helper 2 cells and promotes an interleukin 9-producing subset.
Nat Immunol. 9:1341–1346. 2008.
|
|
65
|
Kaplan MH, Daniel C, Schindler U and
Grusby MJ: STAT proteins control lymphocyte proliferation by
regulating p27Kip1 expression. Mol Cell Biol. 18:1996–2003.
1998.PubMed/NCBI
|
|
66
|
Zhu J, Guo L, Min B, Watson CJ, Hu-Li J,
Young HA, Tsichlis PN and Paul WE: Growth factor independent-1
induced by IL-4 regulates Th2 cell proliferation. Immunity.
16:733–744. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kaplan MH, Wurster AL, Smiley ST and
Grusby MJ: STAT6-dependent and -independent pathways for IL-4
production. J Immunol. 163:6536–6540. 1999.PubMed/NCBI
|
|
68
|
Bruns HA, Schindler U and Kaplan MH:
Expression of a constitutively active STAT6 in vivo alters
lymphocyte homeostasis with distinct effects in T and B cells. J
Immunol. 170:3478–3487. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kaplan MH, Whitfield JR, Boros DL and
Grusby MJ: Th2 cells are required for the Schistosoma mansoni
egg-induced granulomatous response. J Immunol. 160:1850–1856.
1998.PubMed/NCBI
|
|
70
|
Wurster AL, Rodgers VL, White MF,
Rothstein TL and Grusby MJ: Interleukin-4-mediated protection of
primary B cells from apoptosis through STAT6-dependent
up-regulation of Bcl-xL. J Biol Chem. 277:27169–27175. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Takeda K, Kamanaka M, Tanaka T, Kishimoto
T and Akira S: Impaired IL-13-mediated functions of macrophages in
STAT6-deficient mice. J Immunol. 157:3220–3222. 1996.PubMed/NCBI
|
|
72
|
Martinez FO, Helming L and Gordon S:
Alternative activation of macrophages: an immunologic functional
perspective. Annu Rev Immunol. 27:451–483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Huber S, Hoffmann R, Muskens F and
Voehringer D: Alternatively activated macrophages inhibit T-cell
proliferation by STAT6-dependent expression of PD-L2. Blood.
116:3311–3320. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Szanto A, Balint BL, Nagy ZS, Barta E,
Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, et al: STAT6
transcription factor is a facilitator of the nuclear receptor
PPARgamma-regulated gene expression in macrophages and dendritic
cells. Immunity. 33:699–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yao Y, Li W, Kaplan MH and Chang CH:
Interleukin (IL)-4 inhibits IL-10 to promote IL-12 production by
dendritic cells. J Exp Med. 201:1899–1903. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Furqan M, Mukhi N, Lee B and Liu D:
Dysregulation of JAK-STAT pathway in hematological malignancies and
JAK inhibitors for clinical application. Biomark Res. 1:52013.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bito T, Sumita N, Ashida M, Budiyanto A,
Ueda M, Ichihashi M, Tokura Y and Nishigori C: Inhibition of
epidermal growth factor receptor and PI3K/Akt signaling suppresses
cell proliferation and survival through regulation of Stat3
activation in human cutaneous squamous cell carcinoma. J Skin
Cancer. 2011:8745712011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Jeres A, Clemente MJ, Makishima H, Koskela
H, Leblanc F, Peng Ng K, Olson T, Przychodzen B, Afable M,
Gomez-Segui I, et al: STAT3 mutations unify the pathogenesis of
chronic lymphoproliferative disorders of NK cells and T-cell large
granular lymphocyte leukemia. Blood. 120:3048–3057. 2012.
View Article : Google Scholar
|
|
79
|
Hazan-Halevy I, Harris D, Liu Z, Liu J, Li
P, Chen X, Shanker S, Ferrajoli A, Keating MJ and Estrov Z: STAT3
is constitutively phosphorylated on serine 727 residues, binds DNA,
and activates transcription in CLL cells. Blood. 115:2852–2863.
2010. View Article : Google Scholar
|
|
80
|
Ding BB, Yu JJ, Yu RY, Mendez LM,
Shaknovich R, Zhang Y, Cattoretti G and Ye BH: Constitutively
activated STAT3 promotes cell proliferation and survival in the
activated B-cell subtype of diffuse large B-cell lymphomas. Blood.
111:1515–1523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ritz O, Guiter C, Castellano F, Dorsch K,
Melzner J, Jais JP, Dubois G, Gaulard P, Moller P and Leroy K:
Recurrent mutations of the STAT6 DNA binding domain in primary
mediastinal B-cell lymphoma. Blood. 114:1236–1242. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kis LL, Gerasimcik N, Salamon D, Persson
EK, Nagy N, Klein G, Severinson E and Klein E: STAT6 signaling
pathway activated by the cytokines IL-4 and IL-13 induces
expression of the Epstein-Barr virus-encoded protein LMP-1 in
absence of EBNA-2: implications for the type II EBV latent gene
expression in Hodgkin lymphoma. Blood. 117:165–174. 2011.
View Article : Google Scholar
|
|
83
|
Bromberg JF, Wrzeszczynska MH, Devgan G,
Zhao Y, Pestell RG, Albanese C and Darnell JE Jr: STAT3 as an
oncogene. Cell. 98:295–303. 1999. View Article : Google Scholar
|
|
84
|
Bowman T, Garcia R, Turkson J and Jove R:
STATs in oncogenesis. Oncogene. 19:2474–2488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Haura EB, Turkson J and Jove R: Mechanisms
of disease: Insights into the emerging role of signal transducers
and activators of transcription in cancer. Nat Clin Pract Oncol.
2:315–324. 2005. View Article : Google Scholar
|
|
86
|
Frank DA, Mahajan S and Ritz J: B
lymphocytes from patients with chronic lymphocytic leukemia contain
signal transducer and activator of transcription (STAT) 1 and STAT3
constitutively phosphorylated on serine residues. J Clin Invest.
100:3140–3148. 1997. View Article : Google Scholar
|
|
87
|
Mora LB, Buettner R, Seigne J, Diaz J,
Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, et
al: Constitutive activation of STAT3 in human proSTATe tumors and
cell lines: direct inhibition of STAT3 signaling induces apoptosis
of proSTATe cancer cells. Cancer Res. 62:6659–6666. 2002.PubMed/NCBI
|
|
88
|
Diaz N, Minton S, Cox C, Bowman T, Gritsko
T, Garcia R, Eweis I, Wloch M, Livingston S, Seijo E, et al:
Activation of STAT3 in primary tumors from high-risk breast cancer
patients is associated with elevated levels of activated SRC and
survivin expression. Clin Cancer Res. 12:20–28. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Scholz A, Heinze S, Detjen KM, Peters M,
Welzel M, Hauff P, Schirner M, Wiedenmann B and Rosewicz S:
Activated signal transducer and activator of transcription 3
(STAT3) supports the malignant phenotype of human pancreatic
cancer. Gastroenterology. 125:891–905. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Eifan AO, Furukido K, Dumitru A, Jacobson
MR, Schmidt-Weber C, Banfield G, Durham SR and Nouri-Aria KT:
Reduced T-bet in addition to enhanced STAT6 and GATA3 expressing T
cells contribute to human allergen-induced late responses. Clin Exp
Allergy. 42:891–900. 2012.
|
|
91
|
Hadjur S, Bruno L, Hertweck A, Cobb BS,
Taylor B, Fisher AG and Merkenschlager M: IL4 blockade of inducible
regulatory T cell differentiation: the role of Th2 cells, Gata3 and
PU.1. Immunol Lett. 122:37–43. 2009. View Article : Google Scholar : PubMed/NCBI
|