Introduction
Smad proteins are intracellular mediators of
transforming growth factor-β (TGF-β) and bone morphogenetic protein
signaling pathways necessary for the regulation of a variety of
critical processes, including embryonic development, fibrosis,
tumor development, immune function and wound healing (1,2).
Genomic sequencing has revealed numerous defects in the TGF-β
signaling pathway of human pancreatic cancers (3). Smad4, also known as DPC4 (deleted in
pancreatic carcinoma, locus 4), was first isolated and identified
in pancreatic cancer on human chromosome 18q21.1 (4). Hahn et al reported that ~90% of
human pancreatic cancers show allelic loss at chromosome 18q.
Deletion of chromosome 18q, which encompasses the Smad4 region,
significantly affects the prognosis of pancreatic cancer (4). Individuals with pancreatic cancer
positive for Smad4 have shown a higher survival rate compared with
those with pancreatic cancers negative for Smad4 (5). These findings indicate that the
presence of Smad4 is critical in the development and treatment of
human pancreatic cancers.
Chromosomal deletions occur in a number of cancers
causing either the absence of the gene or loss of its function
(6). According to the National
Human Genomic Research Institute and the National Center for
Biotechnology Information, translocation is defined as a type of
chromosomal abnormality in which a chromosome breaks and a portion
of it reattaches to a different chromosome. Studies have shown that
>90% of human cancers possess a certain type of clonal
cytogenetic change (6–8). The most widely described chromosomal
abnormality involving chromosomal translocation is the Philadelphia
chromosome (9,10). Produced from the fusion of
chromosome 9 and a truncated chromosome 22, the Philadelphia
chromosome leads to an oncogenic BCR-ABL gene, which is responsible
for the development of chronic myelogenous leukemia (11–14).
Thus, chromosomal translocations can lead to the formation of new
genes caused by the merging or ablation of existing genes that
contribute to the oncogenic phenotype.
Characterizing translocated genes involves the use
of banding techniques originally described by Rowley in 1973
(10). However, studies used to
identify homozygous deletions on chromosome 18q in pancreatic
cancer cells utilized PCR-based assays that focused mainly on
characterizing a specific region of chromosome 18q where homozygous
deletions commonly occur, 18q21.1 (4,6). While
deletion mapping can identify missing areas of a chromosome, in the
case of Smad4 in BxPC3 cells, it does not take into consideration
deletion due to translocation. While investigating the synthetic
lethal interactions between the inhibition of as mammalian target
of rapamycin complex 1 (mTORC1) and TGF-β signaling pathways in
Smad4-null BxPC3 cells, the expression of a Smad4-like protein was
identified. The present study therefore investigated whether the
Smad4 gene is actually present in BxPC3 cells, a pancreatic cancer
cell line widely used to represent a Smad4-null genotype.
Materials and methods
Cell lines and cell culture
conditions
The BxPC3 and Panc1 cells used in this study were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA) and were maintained in Roswell Park Memorial Institute
(RPMI) medium and Dulbecco’s modified Eagle’s medium (DMEM),
respectively, supplemented with 10% fetal bovine serum (Hyclone,
Waltham, MA, USA). BxPC3 cells were also obtained from a stock
maintained by Dr Murray Korc (University of Indiana, Indianapolis,
IN, USA). For transfection of siRNA, the cells were plated at a
density of 105 cells/60-mm plate 24 h prior to
transfection. All transfections were performed using Lipofectamine
2000 (Gibco-BRL, Carlsbad, CA, USA) according to the manufacturer’s
instructions.
Materials
Rapamycin was obtained from LC Laboratories (Woburn,
MA, USA). The phosphatidylinositol-3-kinase (PI3K) inhibitor,
LY294002, and Wortmannin were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Total Smad2 (product number
5339S, monoclonal rabbit IgG), total Smad3 (product number 9523P,
monoclonal rabbit IgG) and Smad4 (product number 9515, monoclonal
rabbit IgG) primary antibodies were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA), and the glyceraldehyde
3-phosphate dehydrogenase antibody was obtained from Santa Cruz
Biotechnology Inc. (Santa Cruz, CA, USA). Smad4 siRNA and
non-targeted negative control siRNA duplexes were obtained from
Santa Cruz Biotechnology Inc. Smad4 primers were designed and
synthesized by IDT (Coralville, IA, USA). A DNeasy Blood and Tissue
kit was obtained from Qiagen (Hilden, Germany) and a Phusion High
Fidelity DNA Polymerase kit was obtained from New England Biolabs
(Ipswich, MA, USA).
Western blot analysis
Proteins were extracted from cultured cells in
modified radioimmunoprecipitation assay buffer (Upstate
Biotechnology Inc., Lake Placid NY, USA). Equal amounts of protein
were subjected to SDS-PAGE separating gels. Electrophoresed
proteins were then transferred to nitrocellulose and subjected to
western blot analysis as described previously (15). The results of the western blot
analysis were quantified using ImageJ software version 1.47 (NIH,
Bethesda, MD, USA)
PCR
Genomic DNA was extracted from the cells using the
Qiagen DNeasy Blood and Tissue kit. PCR amplification was performed
using primers specific to Smad4 DNA and the Phusion High Fidelity
DNA Polymerase kit was used to determine the presence of Smad4 in
each cell line. Exon 1: Forward primer,
5′-ATGCTCAGTGGCTTCTCGACAAGTTG-3′ and reverse primer,
3′-GGGCTTTTTAAAGCCTCTGCACCAG-5′. Exon 2: forward primer,
5′-CCTTGCAACGTTAGCTGT TGT-3′ and reverse primer,
3′-TGAAGCCTCCCATCCAATGTT CTC-5′. Exon 12: forward primer,
5′-GTTGATGTGGATACT TTTCACACCG-3′ and reverse primer,
3′-CTACCACAAAGC TGGCCTCTACCA-5′.
Smad4 siRNA duplexes
Smad4 siRNA (human) is a pool of three different
siRNA duplexes: Exon from nucleic acids 788-962, with sense,
GCAUCGACAGAGACAUACAtt, and antisense, UGUAUGUCUCUGUCGAUGCtt. Exon
from nucleic acids 2277-3086, with sense, GAUGACUGUUGAUGA AGUAtt,
and antisense, UACUUCAUCAACAGUCAUCtt. Exon from nucleic acids
2277-3086, with sense, CAAGGUUGGUUGCUAAGAAtt and antisense, UUCUUA
GCAACCAACCUUGtt. All sequences are provided in 5′→3′
orientation.
Results
PI3K and mTOR complex 1 (mTORC1)
inhibition induces expression of Smad4 in BxPC3 cells
Defects in TGF-β signaling have been reported for
the majority of pancreatic cancers, with deletions or mutations in
Smad4 being most prevalent (3,4). Smad4
is a member of the Smad family and plays a pivotal role in
mediating the downstream effects of the TGF-β signaling pathway
(16,17). As a tumor suppressor, Smad4
regulates TGF-β-mediated epithelial cell growth or inhibition
(18). Rapamycin has been reported
to activate TGF-β signaling by mediating the nuclear translocation
of activated Smad2/3 in complex with Smad4, while having no effect
on the total protein levels (19).
Rapamycin has also been reported to induce a negative feedback loop
that activates the PI3K signaling pathway, which can block the
expression and activation of Smad3, thus inhibiting the TGF-β
signaling pathway (20). The
present study therefore aimed to determine the effect of rapamycin
on Smad2, Smad3 and Smad4 protein levels in Panc1 and BxPC3 cells.
Data shown in Fig. 1A indicate that
rapamycin alone or in combination with the PI3K inhibitor,
LY294002, does not affect the expression levels of Smad2, 3 or 4 in
Smad4 wild-type Panc1 cells. Sole treatment with rapamycin also
does not affect the Smad2, 3 or 4 protein levels in BxPC3 cells;
expression of Smad4 protein was not expected given the previously
reported absence of Smad4 DNA in BxPC3 cells (4). As expected, Smad4 protein was not
detected in the BxPC3 cells (Fig.
1B, left panel). However, upon dual treatment with rapamycin
and the PI3K inhibitor, LY294002, or Wortmannin in the BxPC3 cells,
a protein recognized by a Smad4 primary antibody was expressed
(Fig. 1B, middle and right panels).
This treatment had no effect on the level of Smad2 or Smad3 in the
BxPC3 cells. These data indicate that under conditions where mTORC1
and PI3K signaling pathways are inhibited, a protein containing a
similar amino acid sequence to that of wild-type Smad4 is expressed
in BxPC3 cells.
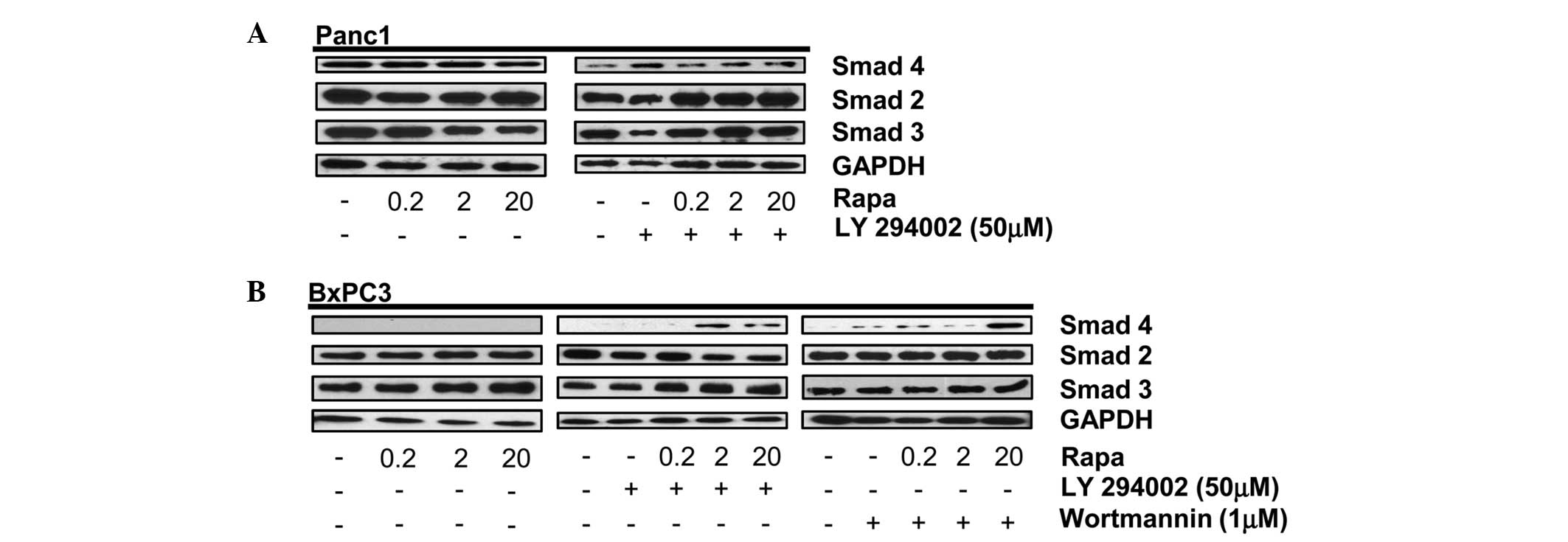 | Figure 1PI3K and mTORC1 inhibition induces
expression of Smad4 in BxPC3 cells. (A) Panc-1 cells were plated at
105 cells/60-mm plate in 10% serum. (A) After 24 h, the
cells were shifted to media containing 10% serum with the indicated
concentration of rapamycin (Rapa) (A, left panel). After another 4
h, the levels of Smad4, Smad2, Smad3 and GAPDH were analyzed by
western blotting. Panc-1 cells were plated at 105
cells/60-mm plate in 10% serum. After 24 h, the cells were shifted
to media containing 10% serum and LY294002 (50 μM) for 1 h (A,
right panel). The cells were then treated with the indicated
concentration of Rapa. After another 4 h, the levels of Smad4,
Smad2, Smad3 and GAPDH were analyzed by western blotting. (B) BxPC3
cells were plated as above and treated with the indicated
concentrations of Rapa for 4 h (left panel) or LY294002 (50 μM;
middle panel) or Wortmannin (1 μM; right panel) for 1 h. Cells
initially treated with LY294002 or Wortmannin were then treated
with the indicated concentration of Rapa. After 4 h, the levels of
Smad4, Smad2, Smad3 and GAPDH were analyzed by western blotting.
Western blots are representative of at least three independent
experiments. PI3K, phosphatidylinositol-3-kinase; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; mTORC1, mammalian target
of rapamycin complex 1. |
siRNA for Smad4 decreases the Smad4-like
protein in BxPC3 cells
The present study showed that Smad4 is detected
under stress conditions in BxPC3 cells (Fig. 1). To further confirm that the
protein expressed upon dual inhibition of the mTORC1 and PI3K
signaling pathways in BxPC3 cells is Smad4, Panc1 and BxPC3 cells
were pretreated with Smad4 siRNA followed by dual treatment with
rapamycin and LY294002. Data in Fig.
2 show that Smad4 siRNA inhibits the expression of Smad4 in
Panc1 cells (Fig. 2A), as well as
the stress-induced Smad4-like protein in BxPC3 cells (Fig. 3B). The human Smad4 siRNA is a pool
of three different siRNA duplexes, one from nucleic acids 788-962
(exon 3) and two from nucleic acids 2277-3086 (exon 12). Therefore,
this data indicates that the abrogated expression of Smad4 in BxPC3
cells is linked to the siRNA duplexes that target either the exon 3
or 12 region of Smad4 DNA. Repeat experiments performed on the
BxPC3 cells obtained from Dr Murray Korc showed the same pattern
(data not shown).
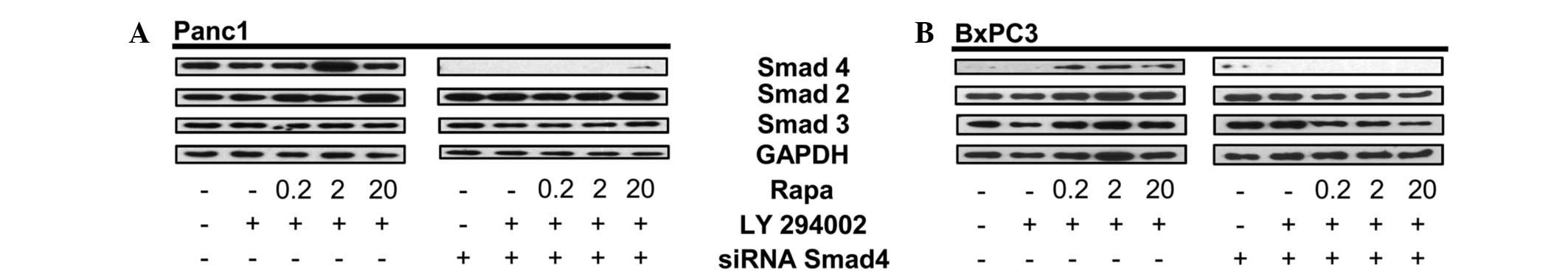 | Figure 2siRNA for Smad4 decreases the
Smad4-like protein in BxPC3 cells. (A) Panc-1 cells were plated in
media containing 10% serum for 24 h. The cells were then
transfected with either control or Smad4 siRNA. After 48 h, the
Panc1 cells were placed in fresh media containing 10% serum and
LY294002 (50 μM) for 1 h, then rapamycin (Rapa; 20 μM) for 4 h at
the indicated concentrations. The total lysate was analyzed by
western blotting for levels of Smad4, Smad2, Smad3 and GAPDH.
Western blots are representative of at least two independent
experiments. (B) BxPC3 cells were plated in media containing 10%
serum for 24 h. The cells were then transfected with either control
or Smad4 siRNA. After 24 h, the BxPC3 cells were placed in fresh
media containing 10% serum and LY294002 (50 μM) for 1 h, then Rapa
(20 μM) for 4 h at the indicated concentrations. The cells were
then collected, lysed and analyzed by western blotting for levels
of Smad4, total Smad2, total Smad3 and GAPDH. Western blots are
representative of at least two independent experiments. GAPDH,
glyceraldehyde 3-phosphate dehydrogenase. |
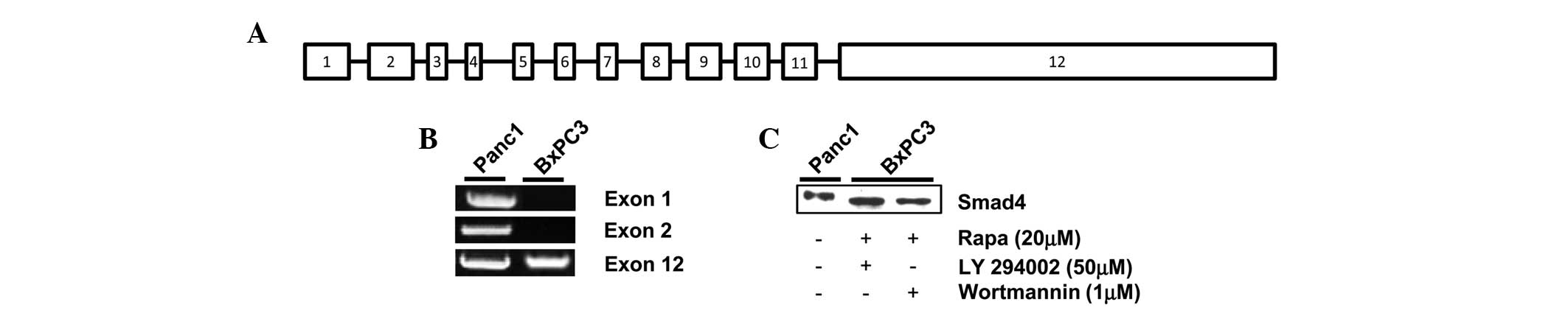
Smad4 DNA is amplified in exon 12 of
BxPC3 pancreatic cancer cells
Proteins are generated via transcription and then
translation from the subsequent DNA sequence. Therefore, proteins
cannot be made without the presence of the corresponding DNA
sequence. It has been previously reported that Smad4 DNA is
amplified in Panc1 but not BxPC3 cells (4,21). It
is important to note that Smad4 DNA contains 12 exons, which encode
for the 552-amino acid Smad4 protein; exon 12 being the largest,
measuring 6.787 kb (Fig. 3A)
(22). Therefore, to determine the
source of the inducible Smad4 protein in the BxPC3 cells, DNA
extracted post-treatment with rapamycin and LY294002 was analyzed
by PCR. Exon 1 of Smad4 has been previously used to identify the
presence or absence of the gene (4). As shown in Fig. 3B, DNA was amplified in the Panc1
cells for exon 1, 2 and 12, the largest primer derived exons, in
the presence of serum. However, when analyzing the BxPC3 cells, a
DNA amplification band was only observed for exon 12 (Fig. 3B). Given that only the Smad4 DNA
fragment corresponding to exon 12 is present in BxPC3 cells, it is
possible that the protein expressed post-treatment with rapamycin
and the PI3K inhibitors is a truncated form of Smad4. As with the
Philadelphia chromosome, depending on the precise translocated
region of the Smad4 gene, the molecular weight of the corresponding
protein can be altered (14).
Therefore, to determine the relative size of Smad4 expressed in the
BxPC3 cells, the cells that were dually treated with high-dose
rapamycin and LY294002 or Wortmannin were analyzed for Smad4 on the
same western blot gel using Panc-1 cells as a positive control. As
shown in Fig. 3C, Smad4 in the
BxPC3 cells had a slightly faster electrophoretic mobility than
wild-type Panc-1 Smad4. Taken together, these data indicate that
BxPC3 cells express an inducible truncated version of the Smad4
protein, encoded mostly in exon 12 of Smad4 DNA.
Discussion
BxPC3 cells are reported to have a homozygous
deletion on chromosome 18q, which encompasses the coding region for
Smad4/DPC4, 18q21.1 (4).
PCR-assays, along with the combination of deletion and physical
mapping, were employed to identify the homozygous deleted regions
of chromosome 18q and resulted in the identification of Smad4 as a
tumor suppressor located at chromosome 18q21.1 (4,6). Hahn
et al reported that chromosomal region 18q21.1 is deleted in
30% of pancreatic cancers, including in BxPC3 cells. Homozygous
deletion of Smad4 has been correlated with the loss of expression
of the corresponding protein (23).
Smad4 plays a pivotal role in regulating TGF-β signaling and can
function as a tumor suppressor or promoter (5,24).
The homozygous loss of Smad4 has been the basis for
use of BxPC3 cells as a model for pancreatic cancer with defective
TGF-β signaling. We previously reported that TGF-β was unable to
rescue BxPC3 cells from rapamycin-induced cell death, as was the
case in the Smad4 wild-type Panc1 pancreatic cancer cells (15) indicating that BxPC3 pancreatic
cancer cells are not responsive to TGF-β. However, in the present
study, we suggest that the basis for defective TGF-β signaling in
BxPC3 cells is not due to the homozygous deletion of Smad4, in that
much of the Smad4 gene is present and expressed in response to the
stress of mTORC1 and PI3K suppression. Thus, it is more likely that
the loss of the Smad4 gene on chromosome 18q is due to a
translocation rather than homozygous deletion. Whether the
truncated Smad4 protein has any phenotypic impact on BxPC3 cells is
not known, however, it appears that BxPC3 cells do not respond to
TGF-β.
Acknowledgements
The authors would like to thank Dr Murray Korc of
the University of Indiana for providing a stock of BxPC3 cells,
purchased from the ATCC, which were used to verify the findings
reported here, and for several helpful suggestions. We are also
thankful to Dr Paul Feinstein of Hunter College, The City
University of New York (New York, NY, USA) for his advice in
pursuing this manuscript and discussions involving genomic mapping
and understanding splicing. This study was supported by a National
Institute of Health grant (R01-CA46677) to DAF, as well as the
Research Centers in Minority Institutions award (RP-03037) from the
National Center for Research Resources of the National Institute of
Health. Further support was provided to individual authors by a
Diversity Supplement (R01-CA46677) and a Minority Biomedical
Research Support-Research and Initiative for Scientific Enhancement
(MBRS-RISE) award.
References
|
1
|
Malkoski SP and Wang XJ: Two sides of the
story? Smad4 loss in pancreatic cancer versus head-and-neck cancer.
FEBS Lett. 586:1984–1992. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moustakas A, Souchelnytskyi S and Heldin
CH: Smad regulation in TGF-beta signal transduction. J Cell Sci.
114:4359–4369. 2001.PubMed/NCBI
|
|
3
|
Jones S, Zhang X, Parsons DW, et al: Core
signaling pathways in human pancreatic cancers revealed by global
genomic analyses. Science. 321:1801–1806. 2008. View Article : Google Scholar
|
|
4
|
Hahn SA, Schutte M, Hoque AT, et al: DPC4,
a candidate tumor suppressor gene at human chromosome 18q21.1.
Science. 271:350–353. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu F: SMAD4/DPC4 and pancreatic cancer
survival. Commentary re: M Tascilar et al, The SMAD4 protein and
prognosis of pancreatic ductal adenocarcinoma Clin Cancer Res 7:
4115–4121, 2001. Clin Cancer Res. 7:3853–3856. 2001.PubMed/NCBI
|
|
6
|
Hahn SA, Hoque AT, Moskaluk CA, et al:
Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer
Res. 56:490–494. 1996.PubMed/NCBI
|
|
7
|
Croce CM: Chromosome translocations and
human cancer. Cancer Res. 46:6019–6023. 1986.PubMed/NCBI
|
|
8
|
Testa JR: Chromosome translocations in
human cancer. Cell Growth Differ. 1:97–101. 1990.PubMed/NCBI
|
|
9
|
Nowell PC and Hungerford DA: Chromosome
studies on normal and leukemic human leukocytes. J Natl Cancer
Inst. 25:85–109. 1960.PubMed/NCBI
|
|
10
|
Rowley JD: Letter: A new consistent
chromosomal abnormality in chronic myelogenous leukaemia identified
by quinacrine fluorescence and Giemsa staining. Nature.
243:290–293. 1973. View
Article : Google Scholar
|
|
11
|
Kurzrock R, Kantarjian HM, Druker BJ and
Talpaz M: Philadelphia chromosome-positive leukemias: from basic
mechanisms to molecular therapeutics. Ann Intern Med. 138:819–830.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Melo JV: The molecular biology of chronic
myeloid leukaemia. Leukemia. 10:751–756. 1996.PubMed/NCBI
|
|
13
|
Talpaz M, Shah NP, Kantarjian H, et al:
Dasatinib in imatinib-resistant Philadelphia chromosome-positive
leukemias. N Engl J Med. 354:2531–2541. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Advani AS and Pendergast AM: Bcr-Abl
variants: biological and clinical aspects. Leuk Res. 26:713–720.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Le Gendre O, Sookdeo A, Duliepre SA, Utter
M, Frias M and Foster DA: Suppression of AKT phosphorylation
restores rapamycin-based synthetic lethality in SMAD4-defective
pancreatic cancer cells. Mol Cancer Res. 11:474–481.
2013.PubMed/NCBI
|
|
16
|
Blobe GC, Schiemann WP and Lodish HF: Role
of transforming growth factor beta in human disease. N Engl J Med.
342:1350–1358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Massagué J, Blain SW and Lo RS: TGFbeta
signaling in growth control, cancer, and heritable disorders. Cell.
103:295–309. 2000.PubMed/NCBI
|
|
18
|
Miyaki M and Kuroki T: Role of Smad4
(DPC4) inactivation in human cancer. Biochem Biophys Res Commun.
306:799–804. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Osman B, Doller A, Akool el S, et al:
Rapamycin induces the TGFbeta1/Smad signaling cascade in renal
mesangial cells upstream of mTOR. Cell Signal. 21:1806–1817. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song K, Wang H, Krebs TL and Danielpour D:
Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated
Smad3 activation. EMBO J. 25:58–69. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Azar R, Alard A, Susini C, Bousquet C and
Pyronnet S: 4E-BP1 is a target of Smad4 essential for
TGFbeta-mediated inhibition of cell proliferation. EMBO J.
28:3514–3522. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang H, Li G, Wu JJ, Wang L, Uhler M and
Simeone DM: Protein kinase A modulates transforming growth
factor-beta signaling through a direct interaction with Smad4
protein. J Biol Chem. 288:8737–8749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wilentz RE, Su GH, Dai JL, et al:
Immunohistochemical labeling for dpc4 mirrors genetic status in
pancreatic adenocarcinomas : a new marker of DPC4 inactivation. Am
J Pathol. 156:37–43. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang G and Yang X: Smad4-mediated TGF-beta
signaling in tumorigenesis. Int J Biol Sci. 6:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|