1. Introduction
Proliferating cell nuclear antigen (PCNA) is a
member of the DNA sliding clamp family, which also includes the
Escherichia coli DNA polymerase (pol) III β-subunit and
phage T4 gene protein (1,2). As a DNA sliding clamp, PCNA with its
high processivity is able to perform the essential tethering of the
replicative DNA pol δ to the template DNA, which is required for
the duplication of an entire genome. The crystal structure analysis
of PCNA reveals a ring-shaped trimeric complex with marked six-fold
symmetry, which encircles the double-stranded DNA that it freely
slides along (3,4). This structural information provides a
vital explanation for the stable association between PCNA and the
cellular DNA without PCNA binding directly to it, as well as by
what means the pol complex links to the DNA strand in a processive
manner.
The proteins that interact with PCNA have been well
mapped (5–7) and one of the most significant
observations that has emerged regarding the investigation of
PCNA-binding proteins is that a number of partners contain a
conserved PCNA-binding motif, the PCNA-interacting protein
(PIP)-box. As predicted, a number of PIPs are involved in various
aspects of DNA replication and processing, possibly through the use
of the sliding clamp properties of PCNA to mediate their
interactions with DNA. Thus, the overlapping nature of the
combining sites for these PCNA binding partners indicated that the
various factors must act sequentially and coordinately to perform
their functions (8). Subsequently,
the conserved PCNA-binding motif may provide a regulatory mechanism
to coordinate different aspects of DNA metabolism, such as the cell
cycle checkpoints, DNA replication and repair.
Previous studies have revealed that PCNA is
ubiquitinated in the response to several DNA damaging agents. This
modification occurs at the Lys164 residue of PCNA (9,10) and
it has been identified that modification of PCNA enhances the
binding of translesion synthesis (TLS) pols, such as pol ι and pol
η, enabling lesion bypass (11).
Although ubiquitinated PCNA is considered to result in the
recruitment of damage tolerance pols to the stalled replication
forks, the definite mechanisms of this modification remain largely
unknown. The present review focuses on the post-translational
modifications of PCNA, and its cellular functions for DNA
replication and repair. Specifically, the important role of
modified PCNA in mediating mammalian cellular response to different
types of DNA damage is highlighted.
2. Modification of PCNA by ubiquitin
(Ub)
Ub is a highly conserved protein with a 76 amino
acid residue polypeptide, and only three amino acid differences
have been identified between the protein in yeast and human cells.
The C-terminus of Ub contains a conserved glycine residue to enable
attachment to substrates, which is activated in an ATP-dependent
manner linked with the cysteine residue of the Ub-activating (E1)
enzyme to form a thiol ester linkage (12). The activated Ub is transferred to
the Ub-conjugating (Ubc) enzymes (E2) to form an additional thiol
ester linkage. Subsequently, and dependent on the aid of a Ub
ligase (E3), the Ub covalently attaches to the lysine residues of
target proteins (13).
To the best of our knowledge, the ubiquitination of
target proteins involves the concerted action of the E1, E2 and E3
enzymes. The E1 and E2 enzymes form thiol ester adducts with Ub and
the two factors are essential for the ubiquitination of substrates.
The majority of organisms possess a single E1, multiple E2 and even
more E3 enzymes (14). Therefore,
the substrates may be modified by a single Ub (termed
monoubiquitination) or by multiple Ubs in the form of an
isopeptide-linked polyUb chain. These various modifications,
particularly mono- versus polyubiquitination, often lead to
qualitatively different outcomes (15). Although the majority of known cases
of Ub modification are focused on targeting substrate proteins to
the proteasome for degradation, studies have shown that
Ub-dependent proteolysis is crucial in the regulation of a number
of other cellular processes, such as cell signaling, cell cycle
progression and DNA repair. Therefore, variation in Ub signaling
may result from the use of different lysine residues in polyUb
chain assembly (16). For example,
the Lys-48-linked chain signals target proteasomes for degradation,
whereas the Lys-63-linked chain constitutes a non-degradative
signal in various pathways, such as DNA damage response and
ribosomal protein synthesis (17).
Previous studies have revealed that PCNA is a major
target for Ub modification during DNA damage response signaling
pathways. The modification of PCNA by a single Ub at the Lys164
residue, in response to DNA damage, is termed monoubiquitination,
which is regulated by the RAD6–RAD18 E2–E3 complex (18). The E2 RAD6 collaborates with E3
RAD18 and attaches the Ub to substrate proteins. The RAD18 gene in
Saccharomyces cerevisiae (S. cerevisiae) belongs to the RAD6
epistasis group, which is highly conserved throughout eukaryotes.
RAD18 contains a middle zinc finger domain, a SAP domain and a
RING-finger domain at its N-terminal. As a key factor in the
postreplication repair (PRR) pathway, RAD18 forms a tight E2–E3
complex with RAD6 to promote PCNA ubiquitination, which in turn is
a crucial cellular regulation mechanism of the PRR pathways that
are conserved in eukaryotes (19–21).
Previous studies have shown that monoubiquitination of PCNA at
Lys164, catalyzed by the RAD6–RAD18 complex, signals for
error-prone repair, possibly by promoting the recruitment of a TLS
pol. Compared with the replicative polymerases, these TLS
polymerases typically contain non-restrictive active sites and lack
3′-5′ proofreading exonuclease activity, which allows them to
accommodate distortions in the DNA (18).
The monoubiquitination of PCNA at the conserved
Lys164 by the RAD6–RAD18 complex has been reported in a wide
variety of organisms. Polyubiquitination of PCNA may be observed in
yeast and mammal cells, however, this modification is also located
at the Lys164 residue. The same Ub modification serves as the
substrate for the extension of a Lys63-linked polyUb chain, which
requires a ternary complex composed of the RING E3 protein, RAD5,
as well as the heterodimeric methyl methanesulfonate (Mms) 2-Ubc13
complex. Ubc13 is a canonical E2 enzyme, however, Mms2 belongs to a
small family of E2 enzyme variant proteins, which resemble E2
enzymes but lack the defining E2 active site cysteine residue
(15,17). The Mms2-Ubc13 complex functions as
an E2 enzyme that is specialized for the assembly of Lys-63-linked
polyubiquitination chains. Based on this genetic evidence, the
modification of PCNA by Lys-63-linked polyUb chains is necessary
for the induction of the error-free DNA repair pathway upon DNA
damage response. The formation of the Lys63-polyUb chains upon the
ubiquitination of PCNA at Lys163 protects cellular DNA against
error-prone TLS-induced genomic mutations, presumably via a
template-switching mechanism using the newly synthesized sister
chromatid as a template to promote the recovery of the blocked
replication forks (17,22). Notably, the same Lys164 residue of
PCNA at which Ub modification occurs has also been identified to be
a target for small Ub-related modifier (SUMO) modifications
(23,24). Furthermore, the modifications by Ub
and SUMO are induced by replication stress or DNA damage and
promote the different branches of DNA damage bypass.
3. Modification of PCNA by SUMO
The SUMO protein shares ~18% of its amino acid
sequence identity with Ub and the two proteins share a similar
three-dimensional structure. The genetic features of SUMO proposed
to influence the interactions of substrates with other proteins or
DNA are considered to be antagonists of the Ub protein. The SUMO
pathway is initiated by a SUMO-activating enzyme termed E1, which
transfers the activated SUMO to the E2 conjugating enzyme, Ubc9.
SUMO is subsequently transferred from Ubc9 to the substrate with
the assistance of the E3 Siz1 pathway (25). Similar to Ub, SUMO conjugates with a
lysine residue to target the substrate (26). However, unlike the target substrates
in ubiquitination for degradation, sumoylation is involved in, and
regulates, numerous cellular metabolism processes, such as
transcriptional regulation, nuclear transport, apoptosis and
protein stability (27).
In yeast, a reciprocal pull-down experiment using
wild-type PCNA and mutant PCNA demonstrated that PCNA may be
sumoylated at the Lys127 and Lys164 residues. In addition, the
double mutant K127R and K164R of PCNA were demonstrated to disturb
the sumoylation of PCNA below detectable levels (28). Lys127 is a hydrophobic residue,
which has been postulated to be a SUMO modification consensus site
and confirmed as a unique component of yeast PCNA (29). By contrast, the Lys164 residue of
PCNA is the major modification site of SUMO and Ub, which is
commonly observed between yeast and humans. The two residues are
located on the outside rim of the trimeric PCNA ring and are
positioned distally from the encircled DNA (30). Lys127 is located in a large loop of
PCNA, which connects the two adjacent terminal domains of the PCNA
monomer that subsequently mediate polymerase interaction with the
connecting loop (31,32). This indicates that the SUMO
modification of PCNA at this site may interfere with DNA polymerase
binding to PCNA. Currently, it is hypothesized that the prominent
binding site for SUMO protein conjugation is the Lys164 residue,
which is conserved within yeast and mammals. A previous study
demonstrated that when the Lys164 residue of PCNA is experimentally
mutated the SUMO conjugation at the Lys127 residue is stimulated,
indicating that modifications by PCNA and SUMO occur on the same
molecule (33).
Biochemical investigations have demonstrated an
association between ubiquitination and sumoylation (34). As the two modifiers compete for
binding at the same lysine residue, studies have hypothesized that
sumoylation at the Lys164 residue of PCNA may function as an
antagonist for Ub proteins (35).
However, additional studies have revealed that the sumoylation of
PCNA at the Lys164 residue does not merely interfere with Ub
modification, but appears to be involved in other functions with
the substrates. As ubiquitination and sumoylation are reversible
processes (26,36), a sequential regulation of
ubiquitination and sumoylation for substrates may be predicted.
Therefore, it appears reasonable to presume that the SUMO and Ub
transforming mechanism may be a prevalent event for the cellular
metabolism process.
Thus far, studies have provided evidence that PCNA
monoubiquitination is required for error-prone TLS. PCNA K63-linked
polyubiquitination governs the template switch-dependent
replication of DNA lesions via an error-free pathway, whereas the
modification of PCNA by SUMO prevents recombination and regulates
the template switch (37). In S.
cerevisiae, the helicase Srs2 is recruited via a conserved
SUMO-interaction motif in the C-terminus of the Srs2 by sumoylated
PCNA (38). The recruitment of DNA
helicase Srs2 disrupts RAD51 single-stranded presynaptic filaments,
thereby interfering with the homologous recombination (HR)
(37,39). Furthermore, previous studies have
shown that the expression of the Lys164 site in mutant PCNA leads
to the increased formation of double-strand breaks (DSBs) in the
RAD18(−/−) cell line where the effect of the RAD18-dependent Lys164
PCNA ubiquitination can be ruled out. In addition, the expression
of the PCNA-SUMO fusion prevents DSB formation and inhibits
recombination as a result of replication stalling at DNA lesions
(40,41). These observations demonstrate the
importance of the SUMO modification of human PCNA in preventing
replication-fork collapse at DSBs and providing genome
stability.
4. PCNA ubiquitination in PRR
PRR was first identified as a means for the repair
of single-stranded gaps during the DNA replication process that is
induced by ultraviolet (UV) light damage. In wild-type cells, PRR
is accomplished by at least two downstream pathways with completely
distinct biological outcomes. The first is the TLS pathway, which
involves a number of non-classical DNA polymerases, such as the
Y-family of DNA polymerases, which bypass specific DNA damage
lesions using the unrepaired DNA strand as a template. The second
pathway is proposed to involve a template switch mechanism,
dependent on RAD5 and the Mms2-Ubc13 complex, which are considered
to allow extension by transiently pairing the blocked nascent
strand and the bypass of DNA damage via the recruitment of HR
machinery (18,42). As TLS uses damaged DNA as a template
and recruits low fidelity Y-family DNA polymerases (that frequently
incorporate incorrect nucleotides during replication of the DNA
damage site), the process is considered to be an error-prone
pathway of DNA synthesis. By contrast, the template switch
mechanism, which is considered to be a relatively error-free
pathway, utilizes the newly synthesized sister chromatid as a
template (43,44).
DNA synthesis by classical polymerases is frequently
blocked by a variety of lesions, however, these replication
blockages can be overcome by PRR pathways. In TLS, the stalled
classical, replicative polymerase is replaced by a non-classical
polymerase that is capable of replicating past the lesion. A
previous study analyzing a yeast model clearly demonstrated the
significant role of the PRR pathway in maintaining genomic
stability (45). In addition,
several lines of evidence have revealed that PCNA is pivotal for
initiating and selecting the different bypass modes of PRR. In
yeast, DNA damage, which is induced by the monoubiquitination of
PCNA at the Lys164 residue, is mediated by the RAD6–RAD18 complex
at a stalled DNA replication fork (9). PCNA monoubiquitination may also
trigger the replacement of replicative polymerases with
non-classical TLS polymerases, which are able to replicate past the
DNA lesions. Furthermore, the Ub binding motif in the majority of
TLS polymerases and/or the PCNA interaction motif appear to be
significant in the regulation of the TLS pathway. The TLS pathway
uses low fidelity DNA polymerases that usually repair the DNA in an
error-prone manner since these polymerases have no proofreading
activity. Several mammalian and yeast TLS polymerases have been
completely identified, including pol η, REV1, REV3, pol ι and pol
κ. These low fidelity polymerases allow replication past a variety
of DNA lesions without repairing the damage (46). In addition, PCNA is further
polyubiquitinated by the RAD5 and Ubc13-Mms2 pathways, which add a
non-canonical Lys-63-linked polyUb chain onto the monoubiquitinated
Lys164 residue of PCNA. Once modified by the polyUb chain, PCNA
triggers TLS using a vague template switch mechanism, which
involves the utilization of specific HR proteins and newly
synthesized sister chromatids to bypass the DNA damage in an
error-free manner. However, the synthesis achieved by these
damage-tolerant polymerases remains controversial in higher
eukaryotes (47). Furthermore, the
sumoylation of PCNA at the Lys164 residue has been found to inhibit
the template switch pathway. This antagonistic effect occurs as the
sumoylated PCNA recruits a DNA helicase, termed Srs2 (23), which disrupts RAD51 nucleoprotein
filaments that are fundamental to the initiation of HR.
Eukaryotes possess several low fidelity DNA
polymerases, which differ from the classical polymerases in their
ability to regulate damaged DNA templates. For example, the
Y-family DNA pol η functions in the error-free TLS of the
UV-induced formation of thymine dimers. By contrast, the DNA pol ζ
functions in mutagenic TLS to bypass DNA lesions (48,49).
At present, the best understood polymerase that is involved in TLS
and tumorigenesis is pol η and the lack of pol η in humans results
in a cancer-prone genetic disorder, the variant form of xeroderma
pigmentosum. In mammal cells, pol η, Rev1, pol ι and ubiquitinated
PCNA colocalize to the replication foci following DNA damage
(50). In addition, in wild-type
cells, pol η specifically interacts with the ubiquitinated PCNA
following DNA damage, however, not with the unmodified PCNA. Thus,
in the presence of ubiquitinated PCNA, the classical DNA pol δ on
the DNA may be replaced by pol η when the replication fork stalls
at the damaged DNA in vivo (51). The two branches of the PRR pathway,
the error-free and the highly mutagenic branches, are likely to
maintain a dynamic balance in cells. However, these branches are
defective in the error-free PPR pathway of yeast cells and,
therefore, spontaneous mutation rates may be elevated by 30-fold
(52), which may be considered to
be a cancer predisposition factor. Ubiquitinated PCNA mediates
error-prone DNA synthesis, which has been postulated as a primary
factor for genomic instability and cancer development, although,
the direct evidence is minimal. Thus, PRR, which is a process that
is orchestrated by ubiquitinated PCNA, appears to be critical for
DNA damage tolerance.
5. PCNA ubiquitination in DSBs
DSBs are the most severe cytotoxic form of DNA
damage, generated by ionizing radiation (IR), mechanical stress on
chromosomes, radiomimetic chemicals, such as camptothecin (CPT), or
the encounter of other types of DNA lesions by the replication
machinery (53,54). As one of the most lethal forms of
DNA damage, if repaired incorrectly or left unrepaired, DSBs result
in chromosomal instability, which eventually leads to cell death or
cancer genesis. DSBs are repaired by the HR pathway, which uses the
newly synthesized sister chromatid as a template or by the
non-homologous end-joining (NHEJ) pathway, which directly joins the
broken DNA ends (55). Increasing
evidence has implied that, in addition to its traditional functions
to bypass DNA damage, ubiquitinated PCNA also functions in
repairing DSBs in vertebrae. However, the exact manner in which
ubiquitinated PCNA is involved in the DSB repair process remains
unknown.
Previous in vitro and in vivo studies
have revealed that PCNA ubiquitination may be activated as a result
of multiple types of incidents. In mammal cells, studies have
revealed that a wide range of DNA damage agents trigger PCNA
ubiquitination, including alkylating agents (such as Mms), bulky
adducts (for example, benzo(a)pyrene diol epoxide [BPDE]),
crosslinking agents (such as cisplatin) and photoproducts induced
by UV irritation (56–59). Previous studies have also indicated
that modified PCNA observed at the blocked DNA replication forks or
replication-independent events, such as DSBs induced by bleomycin
and IR-induced DSBs that are not accompanied by base damage, do not
trigger the ubiquitination of PCNA (60,61).
Furthermore, in budding yeast, treatment with the topoisomerase
inhibitor, CPT, which results in DNA replication fork stalling and
even breakdown at the DNA damaged site, does not active PCNA
ubiquitination (62). The results
of a recent study showed that the modification of PCNA is clearly
induced in budding and fission yeast following treatment with DSB
mutagenic agents, IR or homothallic switching (HO) endonuclease.
However, in mammalian cells treated with IR, PCNA ubiquitination
was not detected. Therefore, further investigations are required to
provide satisfactory explanations for these discrepancies (63). Although, data exists showing that
DSBs induce PCNA ubiquitination, the different modes of DNA damage
response mechanisms that are regulated by the PCNA ubiquitination
pathway remain enigmatic.
Recent studies on budding yeast have reported that
the use of agents to generate pure DSBs, such as HO endonuclease,
induce the PCNA-REV1 interaction, which is mediated by the
ubiquitinated PCNA. In addition, following the generation of pure
DSBs induced by HO expression, the RAD6–RAD18 complex-mediated PCNA
ubiquitination activates the Rev1- and pol ζ-dependent DSB repair
pathways (64). The possible
mechanism of this action may be due to a lack of NHEJ activity. As
the simple rejoining of damaged DNA ends via the NHEJ pathway does
not occur, the DSBs are processed by exonuclease activities to
generate ssDNA tracts at the DSB ends. The new ssDNA tracts may
generate gaps with 3′-termini upon which the PCNA is loaded by
replication factor C (65). Once
loaded, the PCNA is ubiquitinated by the RAD6–RAD18 pathway and in
turn, the PCNA ubiquitination may stimulate the activities of
nearby Rev1 or pol ζ. Thus, the RAD6–RAD18 and Rev1-pol ζ complexes
accumulate at sites close to the DSB ends (64,66).
PCNA ubiquitination may, therefore, provide a direct platform for
the activation of TLS polymerases, pol ζ and Rev1, which are
essential for the DSB repair pathway.
6. PCNA ubiquitination in the cell cycle
checkpoint pathways
The cell cycle checkpoints are signal transduction
pathways that respond to damaged DNA by inhibiting cell cycle
progression (67). The cell cycle
checkpoints also control the fidelity of eukaryotic cell division,
by controlling the orderly progression of critical cell cycle
events, such as DNA replication and chromosome segregation, as well
as ensuring the proper repair of damaged DNA. The cell cycle
delays, that are elicited by the checkpoint signaling pathways,
enable the integration of cell cycle progression with DNA repair.
Consequently, the cell cycle checkpoints are important for
preserving the integrity of the genome.
Acting as one of the significant regulatory
mechanisms responsible for sensing DNA replication stress and
damage, the DNA replication-dependent S phase checkpoint is
considered to be important for guarding the stabilization of
stalled replication forks. The S phase checkpoint, known as a
surveillance system that prevents the firing of late replication
origins, controls chromosome replication, prevents cell-cycle
progression to mitosis, and is important for detecting and
responding to DNA damage and repair (68,69).
Furthermore, as the modification of PCNA is significant for DNA
replication to bypass DNA damage, the modification of PCNA is
considered to be most relevant during the S phase cell cycle
checkpoint. In addition, previous observations have indicated that
PCNA in budding yeast, consistent with its replicative function in
response to DNA damage, is modified primarily during the S phase,
whereas DNA damage in the G1 or G2 phases does not generally
trigger the ubiquitination of PCNA. This indicates that all
modified PCNA predominantly arise from S phase cells, even in
asynchronous populations (63).
Previous studies have provided evidence to
demonstrate that ubiquitinated PCNA may be detected in haploid G1
cells treated with DNA interstrand cross-link (ICL) agents
(70). The biochemical and genetic
studies indicated that only monoubiquitinated PCNA is induced by
ICL damage, with no detection of polyubiquitination or sumoylation.
The likely explanation for this is that the blocked DNA synthesis,
induced by ICL agents, leads to PCNA monoubiquitination, which may
regulate the exchange of DNA pol δ to the error-prone pol ζ. In G1
cells, mutation of the conserved Lys164 of PCNA to arginine
abrogates the capability of DNA pol ζ to associate with chromatin
following ICL damage. However, the RAD5-Mms2-Ubc13 complex-mediated
polyubiquitination of PCNA at Lys164 may lead to an alternative
error-free template switch model following the generation of a
sister chromatid that is likely to occur in the late S and G2
phases (71). In mammal cells
modified with bulky adducts, for example by using BPDE, the S phase
checkpoint pathway is elicited (72). In addition, when the DNA replication
process encounters BPDE-induced bulky adducts during S phase, the
covalent modification of PCNA actives the exchange of the
replicative polymerases with damage-tolerant enzymes. Briefly, in
yeast and mammalian systems, the RAD6–RAD18 complex mediates the S
phase-dependent monoubiquitination of PCNA, which may lead to the
regulated activation of DNA pol ζ in a DNA damage bypass-dependent
manner (73).
7. Outlook
A number of previous studies have analyzed the
post-translational modifications of PCNA and revealed its
importance in the DNA damage response and maintenance of genomic
integrity. The modifications of PCNA are known to influence the
choice of different pathways for the processing of DNA lesions
during replication (Fig. 1). To the
best of our knowledge, the monoubiquitination of PCNA at Lys164 by
the RAD6–RAD18 complex may function to activate DNA damage
tolerance pathways, whereas further extension of this modification
mediated by RAD5 and the UBC13-MMS2 complex, termed
polyubiquitination, triggers an alternative template switching
mechanism (74–78). PCNA sumoylation also targets the
same residue as that targeted in ubiquitination via the recruitment
of Srs2 during the S phase, which serves to inhibit the HR pathways
at the stalled replication fork.
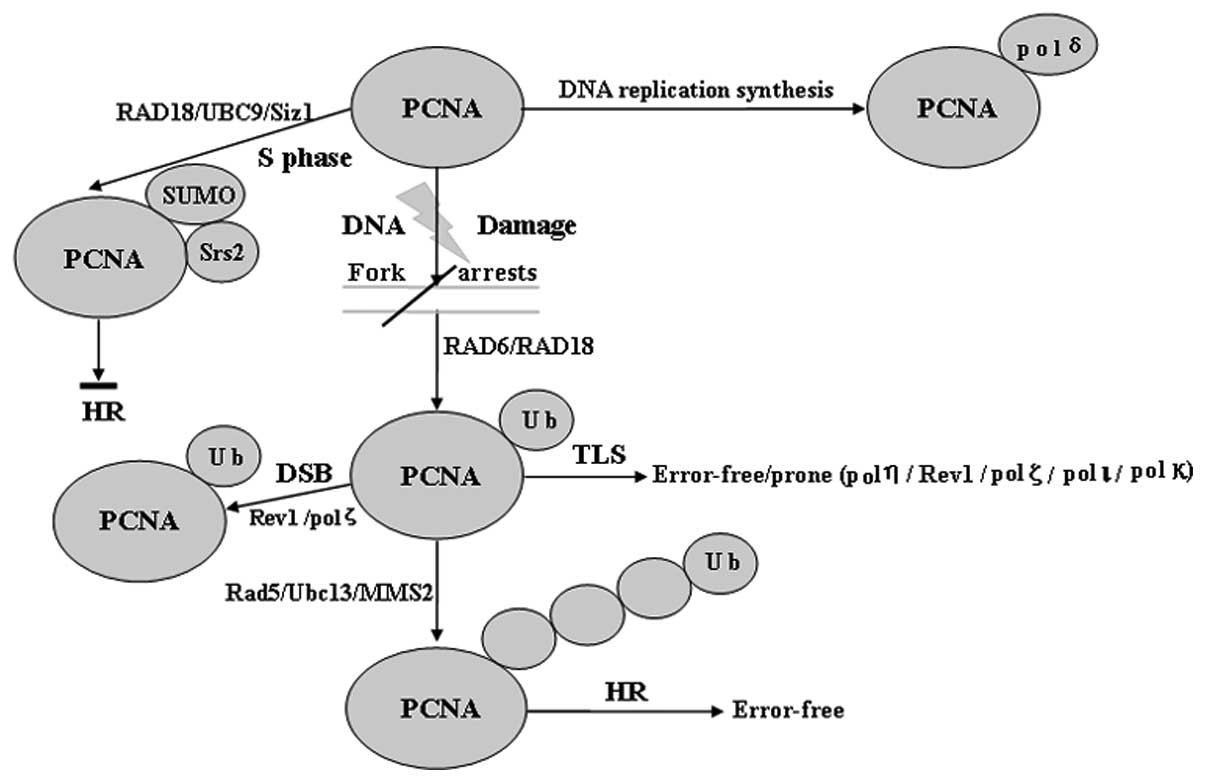 | Figure 1Role of Ub and SUMO modification of
PCNA. PCNA may be modified by monoubiquitination, Lys-63-linked
polyUb chains or SUMO at the same lysine 164 residue. PCNA
monoubiquitination catalyzed by Rad6 and Rad18 directly activates
TLS polymerases (such as pol η, Rev1 and pol ζ), which enable
error-free or error-prone damage bypass, whereas Ubc13/Mms2 and
Rad5 are required to extend the modification by a Lys-63-linked
polyUb chain. PCNA polyubiquitination may occur if TLS fails, which
subsequently results in a recombination-related error-free DNA
damage tolerance pathway. Sumoylation of PCNA occurs in the S phase
and attracts the antirecombinogenic helicase, Srs2, to inhibit
unwanted recombination during DNA synthesis, however,
ubiquitination of PCNA specifically occurs in cells with DNA damage
or stalled replication. SUMO, small ubiquitin-related modifier;
PCNA, proliferating cell nuclear antigen; pol, polymerase; TLS,
translesion synthesis; UBC, ubiquitin-conjugating; DSB,
double-strand break; Ub, ubiquitin; HR, homologous
recombination. |
In addition to its function as a sliding clamp that
ensures the processivity of replicative DNA polymerases, PCNA
serves as a binding platform for the various enzymes involved in
DNA repair, chromatin assembly and cell cycle control (79). In the context of DNA replication and
repair, SUMO and Ub jointly affect the key signal integrator, PCNA,
at the replication fork. In response to DNA-damaging agents, PCNA
is ubiquitinated at the highly conserved Lys164 residue (80). In S. cerevisiae yeast, the
same lysine residue is modified by SUMO during the S phase,
independent of any DNA damage. Therefore, the post-translational
modifications of PCNA regulate the choice of the different modes of
DNA bypass, depending on the species of ubiquitination,
monoubiquitination of PCNA, activation of error-prone TLS and
polyubiquitination, which may mediate an error-free template
switching pathway (81). The
present review discussed the possible regulatory mechanisms that
control PCNA modifications, emphasizing the important role of
modified PCNA during the replication of the DNA template onto which
PCNA is loaded when activating the relevant Ub and SUMO conjugation
factors. In addition, the review identified similarities, as well
as significant variations among different organisms in the
regulation of PCNA modifications.
In conclusion, despite the great advances that have
been made in the understanding of PCNA ubiquitination in the DNA
damage response pathways, a number of questions remain unanswered.
These questions must be investigated in future studies to provide
more detailed insights into the possible mechanisms by which PCNA
ubiquitination and sumoylation function to regulate cell signal
transduction pathways. In addition, further investigation may
highlight the cellular coordination of these various modifications
in the maintenance of cellular genomic integrity. A major challenge
for the future, with regard to the integration of all these
signals, is to develop a coherent model of the orchestration of the
DNA damage response in time and space. An improved understanding of
the effect of the mutual influences that the two relevant
conjugation systems (ubiquitination and sumoylation) exert on each
other is critically important to aid with the investigation of
different post-translational modifiers, which are activated and
utilized in a coordinated manner for the general preservation of
genomic integrity.
Acknowledgements
The authors would like to thank the members of the
laboratory for their valuable comments and suggestions. The present
study was supported by a grant from the National Nature Science
Foundation of China (grant no. 30901220).
References
|
1
|
Jónsson ZO and Hübscher U: Proliferating
cell nuclear antigen: more than a clamp for DNA polymerases.
Bioessays. 19:967–975. 1997.PubMed/NCBI
|
|
2
|
Kelman Z and O’Donnell M: Structural and
functional similarities of prokaryotic and eukaryotic DNA
polymerase sliding clamps. Nucleic Acids Res. 23:3613–3620. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wyman C and Botchan M: DNA replication. A
familiar ring to DNA polymerase processivity. Curr Biol. 5:334–337.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krishna TS, Kong XP, Gary S, et al:
Crystal structure of the eukaryotic DNA polymerase processivity
factor PCNA. Cell. 79:1233–1243. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Warbrick E, Lane DP, Glover DM and Cox LS:
Homologous regions of Fen1 and p21Cip1 compete for binding to the
same site on PCNA: a potential mechanism to co-ordinate DNA
replication and repair. Oncogene. 14:2313–2321. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsurimoto T: PCNA binding proteins. Front
Biosci. 4:D849–D858. 1999. View Article : Google Scholar
|
|
7
|
Dieckman LM, Freudenthal BD and Washington
MT: PCNA structure and function: insights from structures of PCNA
complexes and post-translationally modified PCNA. Subcell Biochem.
62:281–299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maga G and Hubscher U: Proliferating cell
nuclear antigen (PCNA): a dancer with many partners. J Cell Sci.
116:3051–3060. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Watanabe K, Tateishi S, Kawasuji M, et al:
RAD18 guides poleta to replication stalling sites through physical
interaction and PCNA monoubiquitination. EMBO J. 23:3886–3896.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bi X, Barkley LR, Slater DM, et al: RAD18
regulates DNA polymerase kappa and is required for recovery from
S-phase checkpoint-mediated arrest. Mol Cell Biol. 26:3527–3540.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johnson RE, Haracska L, Prakash S and
Prakash L: Role of DNA polymerase zeta in the bypass of a (6-4) TT
photoproduct. Mol Cell Biol. 21:3558–3563. 2001. View Article : Google Scholar
|
|
12
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar
|
|
13
|
Yeh ET, Gong L and Kamitani T:
Ubiquitin-like proteins: new wines in new bottles. Gene. 248:1–14.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pickart CM: Back to the future with
ubiquitin. Cell. 116:181–190. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsui C, Raguraj A and Pickart CM:
Ubiquitin binding site of the ubiquitin E2 variant (UEV) protein
Mms2 is required for DNA damage tolerance in the yeast RAD6
pathway. J Biol Chem. 280:19829–19835. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ulrich HD: Regulating post-translational
modifications of the eukaryotic replication clamp PCNA. DNA Repair
(Amst). 8:461–469. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiu RK, Brun J, Ramaekers C, et al:
Lysine 63-polyubiquitination guards against translesion
synthesis-induced mutations. PLoS Genet. 2:e1162006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andersen PL, Xu F and Xiao W: Eukaryotic
DNA damage tolerance and translesion synthesis through covalent
modifications of PCNA. Cell Res. 18:162–173. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Prakash L: Characterization of
postreplication repair in Saccharomyces cerevisiae and
effects of RAD6, RAD18, rev3 and RAD52 mutations. Mol Gen Genet.
184:471–478. 1981.
|
|
20
|
Lawrence CW and Christensen R: UV
mutagenesis in radiation-sensitive strains of yeast. Genetics.
82:207–232. 1976.PubMed/NCBI
|
|
21
|
Bailly V, Lamb J, Sung P, et al: Specific
complex formation between yeast RAD6 and RAD18 proteins: a
potential mechanism for targeting RAD6 ubiquitin-conjugating
activity to DNA damage sites. Genes Dev. 8:811–820. 1994.
View Article : Google Scholar
|
|
22
|
Brun J, Chiu R, Lockhart K, et al: hMMS2
serves a redundant role in human PCNA polyubiquitination. BMC Mol
Biol. 9:242008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pfander B, Moldovan GL, Sacher M, et al:
SUMO-modified PCNA recruits Srs2 to prevent recombination during S
phase. Nature. 436:428–433. 2005.PubMed/NCBI
|
|
24
|
Haracska L, Torres-Ramos CA, Johnson RE,
et al: Opposing effects of ubiquitin conjugation and SUMO
modification of PCNA on replicational bypass of DNA lesions in
Saccharomyces cerevisiae. Mol Cell Biol. 24:4267–4274. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnson ES: Protein modification by SUMO.
Annu Rev Biochem. 73:355–382. 2004. View Article : Google Scholar
|
|
26
|
Melchior F: SUMO - nonclassical ubiquitin.
Annu Rev Cell Dev Biol. 16:591–626. 2000. View Article : Google Scholar
|
|
27
|
Su HL and Li SS: Molecular features of
human ubiquitin-like SUMO genes and their encoded proteins. Gene.
296:65–73. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matunis MJ: On the road to repair: PCNA
encounters SUMO and ubiquitin modifications. Mol Cell. 10:441–442.
2002. View Article : Google Scholar
|
|
29
|
Johnson ES and Blobel G: Cell
cycle-regulated attachment of the ubiquitin-related protein SUMO to
the yeast septins. J Cell Biol. 147:981–994. 1999. View Article : Google Scholar
|
|
30
|
Ladner JE, Pan M, Hurwitz J and Kelman Z:
Crystal structures of two active proliferating cell nuclear
antigens (PCNAs) encoded by Thermococcus kodakaraensis. Proc
Natl Acad Sci USA. 108:2711–2716. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Amin NS and Holm C: In vivo analysis
reveals that the interdomain region of the yeast proliferating cell
nuclear antigen is important for DNA replication and DNA repair.
Genetics. 144:479–493. 1996.
|
|
32
|
Eissenberg JC, Ayyagari R, Gomes XV and
Burgers PM: Mutations in yeast proliferating cell nuclear antigen
define distinct sites for interaction with DNA polymerase delta and
DNA polymerase epsilon. Mol Cell Biol. 17:6367–6378. 1997.
|
|
33
|
Gali H, Juhasz S, Morocz M, et al: Role of
SUMO modification of human PCNA at stalled replication fork.
Nucleic Acids Res. 40:6049–6059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Müller S, Hoege C, Pyrowolakis G and
Jentsch S: SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell
Biol. 2:202–210. 2001.
|
|
35
|
Ulrich HD, Vogel S and Davies AA: SUMO
keeps a check on recombination during DNA replication. Cell Cycle.
4:1699–1702. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bergink S and Jentsch S: Principles of
ubiquitin and SUMO modifications in DNA repair. Nature.
458:461–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Branzei D and Foiani M: The DNA damage
response during DNA replication. Curr Opin Cell Biol. 17:568–575.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kerscher O: SUMO junction - what’s your
function? New insights through SUMO-interacting motifs. EMBO J.
8:550–555. 2007.
|
|
39
|
Branzei D and Foiani M: RecQ helicases
queuing with Srs2 to disrupt RAD51 filaments and suppress
recombination. Genes Dev. 21:3019–3026. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Santiago A, Godsey AC, Hossain J, et al:
Identification of two independent SUMO-interacting motifs in Daxx:
evolutionary conservation from Drosophila to humans and their
biochemical functions. Cell Cycle. 8:76–87. 2009. View Article : Google Scholar
|
|
41
|
Ting L, Jun H and Junjie C: RAD18 lives a
double life: Its implication in DNA double-strand break repair. DNA
Repair (Amst). 9:1241–1248. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lehmann AR, Niimi A, Ogi T, et al:
Translesion synthesis: Y-family polymerases and the polymerase
switch. DNA Repair (Amst). 6:891–899. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pâques F and Habe JE: Multiple pathways of
recombination induced by double-strand breaks in Saccharomyces
cerevisiae. Microbiol Mol Biol Rev. 63:349–404. 1999.PubMed/NCBI
|
|
44
|
Goldfless SJ, Morag AS, Belisle KA, et al:
DNA repeat rearrangements mediated by DnaK-dependent replication
fork repair. Mol Cell. 21:595–604. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hishida T, Ohya T, Kubota Y, et al:
Functional and physical interaction of yeast Mgs1 with PCNA: impact
on RAD6-dependent DNA damage tolerance. Mol Cell Biol.
26:5509–5517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lehmann AR: Translesion synthesis in
mammalian cells. Exp Cell Res. 312:2673–2676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang H and Lawrence CW: The error-free
component of the RAD6/RAD18 DNA damage tolerance pathway of budding
yeast employs sister-strand recombination. Proc Natl Acad Sci USA.
102:15954–15959. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Washington MT, Johnson RE, Prakash S, et
al: Accuracy of thymine-thymine dimer bypass by Saccharomyces
cerevisiae DNA polymerase eta. Proc Natl Acad Sci USA.
97:3094–3099. 2000.PubMed/NCBI
|
|
49
|
Johnson RE, Prakash S and Prakash L:
Efficient bypass of a thymine-thymine dimer by yeast DNA
polymerase, Poleta. Science. 283:1001–1004. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kannouche PL, Wing J and Lehmann AR:
Interaction of human DNA polymerase eta with monoubiquitinated
PCNA: A possible mechanism for the polymerase switch in response to
DNA damage. Mol Cell. 14:491–500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Freudenthal BD, Gakhar L, Ramaswamy S and
Washington MT: Structure of monoubiquitinated PCNA and implications
for translesion synthesis and DNA polymerase exchange. Nat Struct
Mol Biol. 17:479–484. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Broomfield S, Chow BL and Xiao W: MMS2,
encoding a ubiquitin-conjugating-enzyme-like protein, is a member
of the yeast error-free postreplication repair pathway. Proc Natl
Acad Sci USA. 95:5678–5683. 2010. View Article : Google Scholar
|
|
53
|
Michel B, Ehrlich SD and Uzest M: DNA
double-strand breaks caused by replication arrest. EMBO J.
16:430–438. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ward JF: DNA damage produced by ionizing
RADiation in mammalian cells: identities, mechanisms of formation,
and reparability. Prog Nucleic Acid Res Mol Biol. 35:95–125. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Weinstock DM, Richardson CA, Elliott B and
Jastin M: Modeling oncogenic translocations: distinct roles for
double-strand break repair pathways in translocation formation in
mammalian cells. DNA Repair (Amst). 5:1065–1074. 2006. View Article : Google Scholar
|
|
56
|
Hoege C, Pfander B, Moldovan GL, et al:
RAD6-dependent DNA repair is linked to modification of PCNA by
ubiquitin and SUMO. Nature. 419:135–141. 2002. View Article : Google Scholar
|
|
57
|
Stelter P and Ulrich HD: Control of
spontaneous and damage-induced mutagenesis by SUMO and ubiquitin
conjugation. Nature. 425:188–191. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Davies AA, Huttner D, Daigaku Y, et al:
Activation of ubiquitin-dependent DNA damage bypass is mediated by
replication protein a. Mol Cell. 29:625–636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang XH and Zou L: Dual functions of DNA
replication forks in checkpoint signaling and PCNA ubiquitination.
Cell Cycle. 8:191–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Huttner D and Ulrich HD: Cooperation of
replication protein A with the ubiquitin ligase RAD18 in DNA damage
bypass. Cell Cycle. 7:3629–3633. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chen S, Davies AA, Sagan D and Ulrich HD:
The RING finger ATPase RAD5p of Saccharomyces cerevisiae
contributes to DNA double-strand break repair in a
ubiquitin-independent manner. Nucleic Acids Res. 33:5878–5886.
2005.PubMed/NCBI
|
|
62
|
Frampton J, Irmisch A, Green CM, et al:
Postreplication repair and PCNA modification in Schizosaccharomyces
pombe. Mol Biol Cell. 17:2976–2985. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Daigaku Y, Davies AA and Ulrich HD:
Ubiquitin-dependent DNA damage bypass is separable from genome
replication. Nature. 465:951–955. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hirano Y, Reddy J and Sugimoto K: Role of
budding yeast RAD18 in repair of HO-induced double-strand breaks.
DNA Repair (Amst). 8:51–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Podust VN and Hübscher U: Lagging strand
DNA synthesis by calf thymus DNA polymerases alpha, beta, delta and
epsilon in the presence of auxiliary proteins. Nucleic Acids Res.
21:841–846. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Garg P and Burgers PM: Ubiquitinated
proliferating cell nuclear antigen activates translesion DNA
polymerases eta and REV1. Proc Natl Acad Sci USA. 102:18361–18366.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Branzei D, Vanoli F and Foiani M:
Sumoylation regulates RAD18-mediated template switch. Nature.
456:915–920. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nyberg KA, Michelson RJ, Putnam CW and
Weinert TA: Toward maintaining the genome: DNA damage and
replication checkpoints. Annu Rev Genet. 36:617–656. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
McHugh PJ and Sarkar S: DNA interstrand
cross-link repair in the cell cycle: a critical role for polymerase
zeta in G1 phase. Cell Cycle. 5:1044–1047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Moldovan GL, Pfander B and Jentsch S: PCNA
controls establishment of sister chromatid cohesion during S phase.
Mol Cell. 23:723–732. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bi X, Barkley LR, Slater DM, et al: RAD18
regulates DNA polymerase kappa and is required for recovery from
S-phase checkpoint-mediated arrest. Mol Cell Biol. 26:3527–3540.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Branzei D, Sollier J, Liberi G, et al:
Ubc9- and mms21-mediated sumoylation counteracts recombinogenic
events at damaged replication forks. Cell. 127:509–522. 2006.
View Article : Google Scholar
|
|
74
|
Niu H, Chung WH, Zhu Z, et al: Mechanism
of the ATP-dependent DNA end-resection machinery from
Saccharomyces cerevisiae. Nature. 467:108–111. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Krejci L, Van Komen S, Li Y, et al: DNA
helicase Srs2 disrupts the RAD51 presynaptic filament. Nature.
423:305–309. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Chen J, Bozza W and Zhuang Z:
Ubiquitination of PCNA and its essential role in eukaryotic
translesion synthesis. Cell Biochem Biophys. 60:47–60. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Haracska L, Kondratick CM, Unk I, et al:
Interaction with PCNA is essential for yeast DNA polymerase eta
function. Mol Cell. 8:407–415. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lee KY and Myung K: PCNA modifications for
regulation of post-replication repair pathways. Mol Cells. 26:5–11.
2008.PubMed/NCBI
|
|
79
|
Papouli E, Chen S, Davies AA, et al:
Crosstalk between SUMO and ubiquitin on PCNA is mediated by
recruitment of the helicase Srs2p. Mol Cell. 19:123–133. 2005.
View Article : Google Scholar
|
|
80
|
Schwartz DC and Hochstrasser M: A
superfamily of protein tags: ubiquitin, SUMO and related modifiers.
Trends Biochem Sci. 28:321–328. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Chiu RK, Brun J, Ramaekers C, et al:
Lysine 63-polyubiquitination guards against translesion
synthesis-induced mutations. PLoS Genet. 2:e1162006. View Article : Google Scholar : PubMed/NCBI
|














