Introduction
Obesity, the accumulation of excess fat tissue, is a
risk factor for the development of postmenopausal breast cancer
(1). The molecular changes in
metabolism associated with obesity are thought to contribute to
this phenomenon (2). One of these
molecular changes is increased circulating leptin levels in obese
individuals (3). Leptin is a 16 kDa
peptide hormone predominantly produced by white fat tissue
(4). Its main function is to signal
to the hypothalamus, which in response regulates satiety and energy
expenditure (5). Notably, leptin is
also responsible for the normal formation of the mammary gland in
humans (6), suggesting an
involvement in mammary tissue growth and differentiation, and
potentially malignant transformation. Epidemiologically, leptin
concentrations are higher in patients with breast cancer compared
with healthy individuals, independent of body weight (7). Additionally, leptin receptor (Ob-R)
expression is increased in breast tumor tissue compared with
surrounding tissue (8). Breast
cancer cell lines have also been found to express leptin and Ob-R
(9).
Previous in vitro experiments investigating
the effects of leptin treatment on cell proliferation and tumor
growth have revealed conflicting results. In MDA-MB-231 breast
cancer cells, leptin induced a robust concentration-dependent
increase in proliferation in two independent studies (10,11).
Conversely, in MCF-7 breast cancer cells, leptin treatment
increased (12) and decreased
(11) proliferation. Similarly, in
human epidermal growth factor (HER)-2 overexpressing SK-BR-3 breast
cancer cells, leptin treatment increased proliferation between 5
and 50 ng/ml, but not at 100 ng/ml (11). These controversial findings warrant
the need for further investigation into the effects of leptin on
proliferation in a human breast cancer in vitro cell
system.
Notably, a number of studies investigating the
effect of cytokines on proliferation changes in cell culture models
used concentrations which are several-fold greater than the highest
known physiological concentration, such as insulin treatment
(13,14) or tumor necrosis factor-α treatment
(15,16). However, previous studies
investigating the effect of leptin treatment on proliferation in
breast cancer cells (10,11,17)
did not or only marginally exceeded maximal physiological leptin
concentrations of 100 ng/ml (3).
The highest leptin concentration used in vitro to examine
changes in T47D breast cancer cell proliferation was 1,000 ng/ml.
However, increased cell proliferation was only observed with up to
100 ng/ml leptin (18). Thus, while
previous data appear to indicate a growth inhibitory effect of
leptin at above physiological concentrations, it has not yet been
fully investigated. Therefore, the present study aimed to explore
the effects of supraphysiological leptin concentrations on
proliferation in two breast cancer cell lines.
The present study aimed to investigate the effects
of physiological and supraphysiological levels of leptin (≤1,600
ng/ml) on proliferation in SK-BR-3 and MDA-MB-231 breast cancer
cells, two cell lines representative of HER-2-positive and
basal-type breast cancer subtypes, respectively. The activation of
phosphatidylinositide 3-kinase (PI3K) and mitogen-activated protein
kinase (MAPK) cell signaling pathways and distribution across cell
cycle stages were assessed in the two cell lines following
treatment with 1,600 ng/ml leptin.
Materials and methods
Cell lines
SK-BR-3 and MDA-MB-231 breast cancer cell lines were
purchased from the American Type Culture Collection (Manassas, VA,
USA). The two cell lines were routinely cultured in RPMI-1640
medium (including 25 mM HEPES, 1× Glutamax™; Gibco, Paisley, UK)
supplemented with 10% fetal calf serum (FCS; Pierce Biosciences,
Cramlington, UK), 100 U/ml penicillin and 100 μg/ml streptomycin
(Gibco).
Bromodeoxyuridine (BrdU) proliferation
assay
Cell proliferation was detected using the
Proliferation ELISA kit (Roche Diagnostics GmbH, Penzberg,
Germany). The two cell lines were plated at a density of
5×103 cells/well in 96-well plates with 100 μl/well
growth medium and incubated for 24 h at 37°C. Cells were starved
for 24 h in RPMI-1640 medium without FCS supplementation, and then
treated for 24 or 48 h with 6.25–1,600 ng/ml leptin in replicates
of six in starvation medium. During treatment, the medium was
supplemented with 10 μM BrdU. Cell proliferation was assessed as
previously described (16). The
experiment was repeated for a total of three independent times.
Each experiment had six replicates for each leptin
concentration.
PI3K and MAPK phosphorylation ELISA
Cell-based ELISA Phospho-Akt (S473) Immunoassay and
Phospho-extracellular signal-regulated kinase (ERK)1/ERK2
(T202/Y204) Immunoassay were purchased from R&D Systems
(Abingdon, UK). The cells were plated in a supplied clear bottom,
black-walled, 96-well plate at a density of 5×103
cells/well with 100 μl/well growth medium, and incubated for 24 h
at 37°C. The cells were starved for 24 h as mentioned above and
then treated with 1,600 ng/ml leptin for 5–20 min in duplicates.
Phosphorylation of protein kinase B (Akt) or ERK1/2 was then
assessed as previously described (16). The experiment was repeated for a
total of three independent experiments with two replicates for each
time point in each experiment.
Cell cycle analysis
Changes in the cell distribution across cell cycle
stages were assessed by measuring the DNA content in cells using
flow cytometry following leptin treatment. The DNA-specific dye was
propidium iodide (PI; Sigma-Aldrich, Gillingham, UK). The cells
were plated at a density of 5×105 cells/well in six-well
plates with 3 ml growth medium, and incubated for 24 h at 37°C. The
cells were starved for 24 h, treated with 1,600 ng/ml leptin for 24
h and then harvested, treated and analyzed as described previously
(16).
Statistical analysis
The findings were analyzed for statistical
significance using univariate analysis of variance between the
control and each treatment concentration for cell proliferation
analysis and between the control and each time point in the cell
signaling pathway analysis, followed by Dunnett’s post hoc t-tests.
Differences in the distribution across cell cycle stages between
the control and leptin-treated cells were assessed for each cell
cycle stage using Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Changes in cell proliferation following
leptin treatment
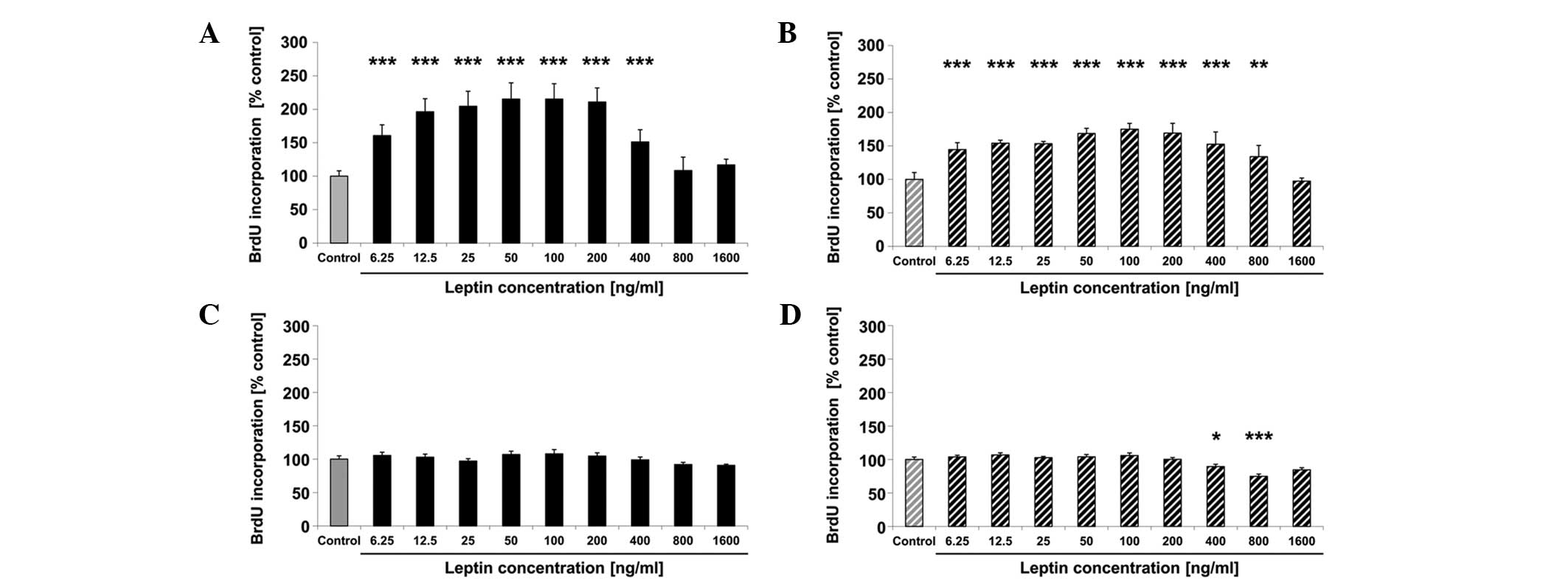
In the SK-BR-3 cells, proliferation increased by 61,
96, 104, 115, 115, 110 and 51% following treatment with 6.25, 12.5,
25, 50, 100, 200 and 400 ng/ml leptin, respectively, for 24 h
compared with the untreated control (all P<0.001) (Fig. 1A). There was no significant
difference in cell proliferation between the untreated cells and
cells treated with 800 or 1,600 ng/ml leptin for 24 h (Fig. 1A). After 48 h of treatment, cell
proliferation increased significantly by 44, 53, 53, 69, 75, 69, 52
and 33% following treatment with 6.25, 12.5, 25, 50, 100, 200, 400
ng/ml (all P<0.001) and 800 ng/ml (P=0.009) leptin, respectively
compared with the untreated control (Fig. 1B). There was no change in
proliferation after 48 h of treatment with 1,600 ng/ml leptin
(Fig. 1B). In MDA-MB-231 breast
cancer cells, proliferation did not change significantly after 24 h
of treatment with leptin (Fig. 1C)
and decreased significantly by 11% (P=0.023) and 26% (P<0.001)
after 48 h of treatment with 400 and 800 ng/ml (P<0.001) of
leptin, respectively, compared with the untreated control (Fig. 1D). Based on the findings obtained on
growth inhibition following treatment with 1,600 ng/ml leptin, cell
signaling and cell cycle changes were assessed to determine the
underlying mechanisms responsible for growth inhibition in the two
breast cancer cell lines.
Changes in PI3K and MAPK cell signaling
pathway activity following leptin treatment
In the SK-BR-3 cells, Akt-phosphorylation did not
change significantly following treatment with 1,600 ng/ml leptin
for up to 20 min compared with the control (Fig. 2A). ERK1/2-phosphorylation decreased
by 32 and 34% after 10 (P<0.001) and 15 min (P<0.001) of
treatment with 1,600 ng/ml leptin, respectively, compared with the
control (Fig. 2B). In the
MDA-MB-231 cells, Akt-phosphorylation did not change significantly
following treatment with 1,600 ng/ml leptin (Fig. 2C), whereas ERK1/2-phosphorylation
decreased significantly by 17 and 20% after 15 (P=0.026) and 20 min
(P=0.011) of treatment with 1,600 ng/ml leptin, respectively,
compared with the control (Fig.
2D).
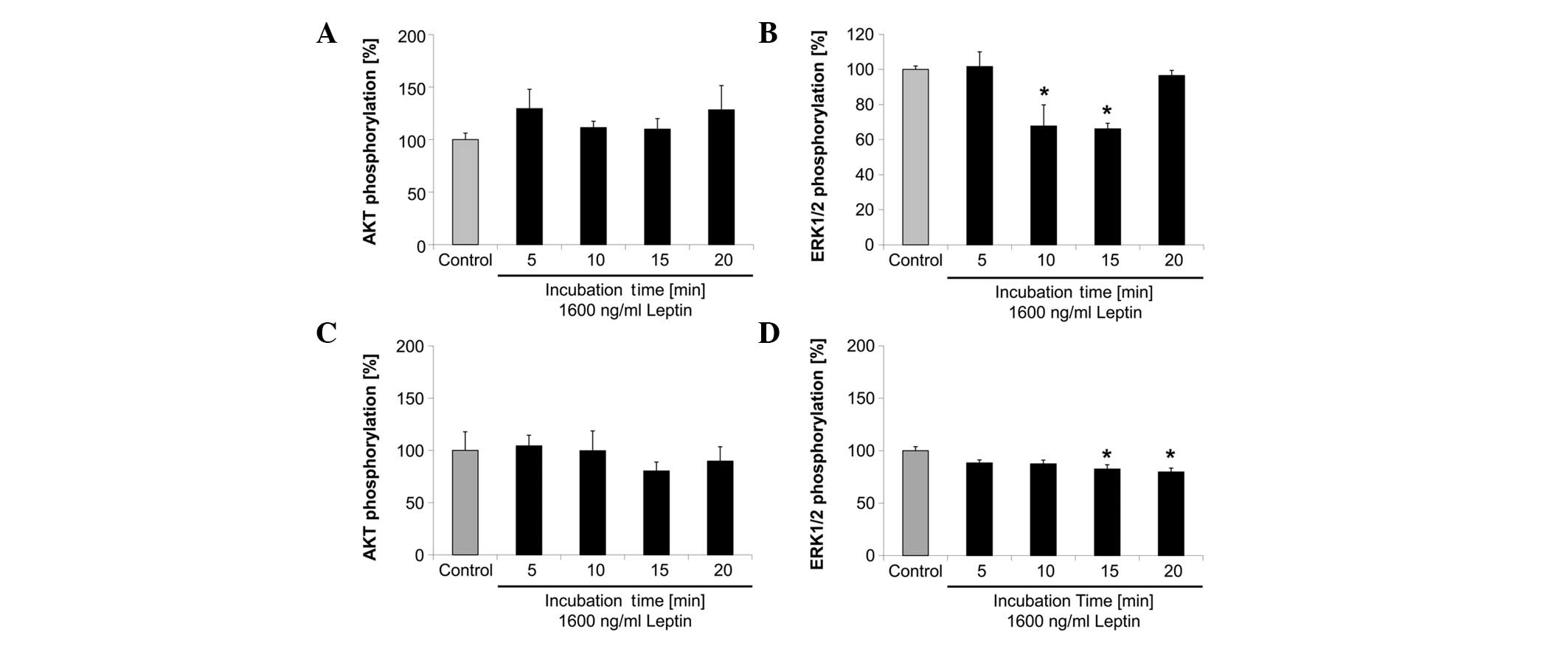 | Figure 2Changes in (A and C) Akt
phosphorylation and (B and D) ERK1/2 phosphorylation as indicators
of changes in the PI3K or MAPK cell signaling pathway,
respectively, following 1,600 ng/ml leptin treatment of (A and B)
SK-BR-3 and (C and D) MDA-MB-231 breast cancer cells for the
indicated time periods. Bars represent Akt or
ERK1/2-phosphorylation in relation to their respective control
within each graph, and are expressed as a percentage of the
control. Error bars represent standard error of the mean of three
experiments, each consisting of two replicates, i.e., six data
points for each bar. Significance was obtained using Dunnett’s post
hoc t-test following univariate analysis of variance
(*P<0.05, vs. the control). ERK, extracellular
signal-regulated kinase; PI3K, phosphatidylinositide-3 kinase;
MAPK, mitogen-activated protein kinase. |
Changes in distribution of cell
population across cell cycle stages
In the SK-BR-3 cells, 1,600 ng/ml leptin treatment
may increase the G1-phase population and decrease the
G2-phase population (Table
I). Cells in the G1-phase increased by 4.5
percentage points (11% increase) and cells in the
G2-phase decreased by 2.0 percentage points (12%
decrease). In MDA-MB-231 cells, the subG1-phase
population increased by 1.2 percentage points (7% increase) and the
G1-phase population decreased by 1.0 percentage point
(2% decrease) following treatment with 1,600 ng/ml leptin (Table I). None of the observed changes were
significantly different.
 | Table IChanges of cell population
distribution across cell cycle stages after 24 h of treatment with
1,600 ng/ml leptin. |
Table I
Changes of cell population
distribution across cell cycle stages after 24 h of treatment with
1,600 ng/ml leptin.
| SK-BR-3 cells | | MDA-MB-231 cells | |
|---|
|
| |
| |
|---|
| Cell cycle stage
(%) | Control | Leptin-treated | P-value | Control | Leptin-treated | P-value |
|---|
| SubG1 | 20.56±3.01 | 20.4±1.68 | 0.2189 | 17.00±1.07 | 18.15±1.33 | 0.5077 |
|
G0/G1 | 39.90±2.00 | 44.385±2.10 | 0.1490 | 55.12±0.97 | 54.14±0.99 | 0.3830 |
| S | 6.30±0.45 | 5.86±0.81 | 0.6444 | 7.23±0.65 | 6.96±0.62 | 0.7677 |
| G2 | 15.98±0.70 | 14.03±0.87 | 0.0863 | 12.88±0.43 | 12.71±0.48 | 0.4257 |
Discussion
Previous in vitro studies have demonstrated
that leptin induces cell proliferation in a variety of breast
cancer cell types, within physiological concentrations (25–100
ng/ml) (6,9–12,18).
Conversely, the same studies did not observe increased cell
proliferation with leptin concentrations exceeding 100 ng/ml. The
findings of the present study confirmed the increased cell
proliferation in SK-BR-3 breast cancer cells, but not in MDA-MB-231
cells at physiological concentrations. Furthermore, leptin
treatment at supraphysiological concentrations did not increase
cell proliferation in the SK-BR-3 cells, but inhibited
proliferation of the MDA-MB-231 cells. To the best of our
knowledge, this study was the first to indicate the potential of
leptin treatment to inhibit cell proliferation in breast cancer
cells. The mechanism by which supraphysiological leptin
concentrations induce growth inhibition may involve decreased
activation of the Ras-mediated MAPK pathway.
As a potential explanation, leptin may interact with
the HER-2/neu receptor in SK-BR-3 breast cancer cells, which is
overexpressed in these cells, resulting in decreased MAPK activity.
Soma et al reported that SK-BR-3 cells treated with leptin
(500 ng/ml) resulted in increased phosphorylation of the HER-2/neu
receptor (20). This cross-talk was
identified as being responsible for an increase in ERK1/2
phosphorylation, which was also observed following leptin
treatment. The findings of the present study suggest that at higher
leptin concentrations, this effect is inhibited. This may either be
by high leptin levels inhibiting the potential of HER-2/neu to
activate ERK1/2 or by inhibiting ERK1/2 phosphorylation directly.
HER-2/neu and Ob-R transduct their proliferative signal through the
PI3K and/or MAPK pathways (21),
suggesting there may be an interaction on the targets downstream of
the two receptors. Thus, leptin may have at least two modes of
action, which appear to be antagonistic. First, leptin increases
phosphorylation of HER-2/neu, which results in increased
proliferation of SK-BR-3 breast cancer cells; second, leptin
inhibits ERK1/2 phosphorylation, which is predominant at high
leptin concentrations, thereby reducing the effect of increased
HER-2/neu signaling.
In MDA-MB-231 breast cancer cells, which are
HER-2-negative, interplay with HER-2/neu cannot account for the
observed reduction in proliferation, indicating that growth
inhibition at supraphysiological leptin concentrations is HER-2/neu
independent. In a study aiming to potentiate the antitumor effects
of cAMP-agonists, leptin induced apoptosis in MDA-MB-231 breast
cancer cells when cAMP levels were increased (22), which resulted in ERK1/2 inactivation
and the subsequent inhibition of protein kinase A (PKA) expression
(23). Thus, at supraphysiological
concentrations, leptin may not require elevated cAMP levels to
decrease PKA, and this may provide a mechanism for the effects
observed in our study.
These findings suggest that leptin exerts a biphasic
effect on cell proliferation in SK-BR-3 breast cancer cells and
that leptin signaling may play a role in breast cancer development
and progression. Therefore, the inhibition of leptin signaling may
be relevant for breast cancer prevention, particularly for obese
individuals showing high levels of leptin and occurrence of breast
cancer. Nutritional interventions (24) or anti-leptin treatment (25) may be considered as a potential
preventative strategy and treatment for HER-2/neu overexpressing
breast tumors, respectively. Further investigations into the
inhibitory effects of leptin at high concentrations may reveal the
unknown mechanisms in the connection between obesity and
postmenopausal breast cancer.
References
|
1
|
Calle EE, Rodriguez C, Walker-Thurmond K
and Thun MJ: Overweight, obesity, and mortality from cancer in a
prospectively studied cohort of U.S. adults. N Engl J Med.
348:1625–1638. 2003.
|
|
2
|
Lorincz AM and Sukumar S: Molecular links
between obesity and breast cancer. Endocr Relat Cancer. 13:279–292.
2006.
|
|
3
|
Considine RV, Sinha MK, Heiman ML, et al:
Serum immunoreactive-leptin concentrations in normal-weight and
obese humans. N Engl J Med. 334:292–295. 1996.
|
|
4
|
Zhang Y, Proenca R, Maffei M, et al:
Positional cloning of the mouse obese gene and its human homologue.
Nature. 372:425–432. 1994.
|
|
5
|
Stephens TW, Basinski M, Bristow PK, et
al: The role of neuropeptide Y in the antiobesity action of the
obese gene product. Nature. 377:530–532. 1995.
|
|
6
|
Hu X, Juneja SC, Maihle NJ and Cleary MP:
Leptin - a growth factor in normal and malignant breast cells and
for normal mammary gland development. J Natl Cancer Inst.
94:1704–1711. 2002.
|
|
7
|
Tessitore L, Vizio B, Pesola D, et al:
Adipocyte expression and circulating levels of leptin increase in
both gynaecological and breast cancer patients. Int J Oncol.
24:1529–1535. 2004.
|
|
8
|
Révillion F, Charlier M, Lhotellier V, et
al: Messenger RNA expression of leptin and leptin receptors and
their prognostic value in 322 human primary breast cancers. Clin
Cancer Res. 12:2088–2094. 2006.
|
|
9
|
Ishikawa M, Kitayama J and Nagawa H:
Enhanced expression of leptin and leptin receptor (OB-R) in human
breast cancer. Clin Cancer Res. 10:4325–4331. 2004.
|
|
10
|
Frankenberry KA, Skinner H, Somasundar P,
McFadden DW and Vona-Davis LC: Leptin receptor expression and cell
signaling in breast cancer. Int J Oncol. 28:985–993. 2006.
|
|
11
|
Ray A, Nkhata KJ and Cleary MP: Effects of
leptin on human breast cancer cell lines in relationship to
estrogen receptor and HER2 status. Int J Oncol. 30:1499–1509.
2007.
|
|
12
|
Dieudonne MN, Machinal-Quelin F,
Serazin-Leroy V, Leneveu MC, Pecquery R and Giudicelli Y: Leptin
mediates a proliferative response in human MCF7 breast cancer
cells. Biochem Biophys Res Commun. 293:622–628. 2002.
|
|
13
|
Costantino A, Milazzo G, Giorgino F, et
al: Insulin-resistant MDA-MB231 human breast cancer cells contain a
tyrosine kinase inhibiting activity. Mol Endocrinol. 7:1667–1676.
1993.
|
|
14
|
Weichhaus M, Broom J, Wahle K and Bermano
G: A novel role for insulin resistance in the connection between
obesity and postmenopausal breast cancer. Int J Oncol. 41:745–752.
2012.
|
|
15
|
Rivas MA, Carnevale RP, Proietti CJ, et
al: TNF alpha acting on TNFR1 promotes breast cancer growth via
p42/P44 MAPK, JNK, Akt and NF-kappa B-dependent pathways. Exp Cell
Res. 314:509–529. 2008.
|
|
16
|
Weichhaus M, Broom I and Bermano G: The
molecular contribution of TNF-alpha in the link between obesity and
breast cancer. Oncol Rep. 25:477–483. 2011.
|
|
17
|
Chen C, Chang YC, Liu CL, Chang KJ and Guo
IC: Leptin-induced growth of human ZR-75-1 breast cancer cells is
associated with up-regulation of cyclin D1 and c-Myc and
down-regulation of tumor suppressor p53 and p21WAF1/CIP1. Breast
Cancer Res Treat. 98:121–132. 2006.
|
|
18
|
Laud K, Gourdou I, Pessemesse L, Peyrat JP
and Djiane J: Identification of leptin receptors in human breast
cancer: functional activity in the T47-D breast cancer cell line.
Mol Cell Endocrinol. 188:219–226. 2002.
|
|
19
|
Okumura M, Yamamoto M, Sakuma H, et al:
Leptin and high glucose stimulate cell proliferation in MCF-7 human
breast cancer cells: reciprocal involvement of PKC-alpha and PPAR
expression. Biochim Biophys Acta. 1592:107–116. 2002.
|
|
20
|
Soma D, Kitayama J, Yamashita H, et al:
Leptin augments proliferation of breast cancer cells via
transactivation of HER2. J Surg Res. 149:9–14. 2008.
|
|
21
|
Reese DM and Slamon DJ: HER-2/neu signal
transduction in human breast and ovarian cancer. Stem Cells.
15:1–8. 1997.
|
|
22
|
Naviglio S, Di Gesto D, Romano M, et al:
Leptin enhances growth inhibition by cAMP elevating agents through
apoptosis of MDA-MB-231 breast cancer cells. Cancer Biol Ther.
8:1183–1190. 2009.
|
|
23
|
Naviglio S, Di Gesto D, Illiano F, et al:
Leptin potentiates antiproliferative action of cAMP elevation via
protein kinase A down-regulation in breast cancer cells. J Cell
Physiol. 225:801–809. 2010.
|
|
24
|
Fan C, Liu X, Shen W, Deckelbaum RJ and Qi
K: The regulation of leptin, leptin receptor and
pro-opiomelanocortin expression by N-3 PUFAs in diet-induced obese
mice is not related to the methylation of their promoters. Nutr
Metab (Lond). 8:312011.
|
|
25
|
Guo S, Liu M, Wang G, et al: Oncogenic
role and therapeutic target of leptin signaling in breast cancer
and cancer stem cells. Biochim Biophys Acta. 1825:207–222.
2012.
|