Introduction
von Hippel-Lindau (VHL) disease is an autosomal
dominantly inherited neoplastic syndrome that is characterized by
hemangioblastomas of the central nervous system (CNS) and retina,
renal cell carcinoma (RCC), pheochromocytoma, pancreatic
neuroendocrine tumors (NETs) and endolymphatic sac tumors (1,2). The
majority of patients are diagnosed following the identification of
CNS tumors. The incidence of VHL disease is ~1 in 36,000 live
births and its penetrance is >90% by the age of 65 years
(3). Tumors of the CNS and RCCs are
the main causes of mortality in VHL patients, accounting for the
median life expectancy of <50 years (4,5).
Well-defined diagnostic criteria have been identified for VHL
disease. If there is a family history, a diagnosis of VHL disease
can be determined by identifying only a single VHL tumor, whereas
if there is no family history, the presence of two VHL tumors is
required for diagnosis (6). VHL
disease is classified into types 1 and 2 according to the absence
or presence of pheochromocytoma, respectively. Type 2 is further
divided into types 2A, 2B and 2C. Type 2A is characterized by a
high risk of hemangioblastoma and pheochromocytoma, with a lower
risk of RCC, and type 2B is characterized by a high risk of
pheochromocytoma, RCC and pheochromocytoma. Cases that present with
only pheochromocytoma are designated as type 2C (6,7).
The VHL gene is localized on the short arm of
chromosome 3, with three exons, and coded for two isoforms of pVHL
(a tumor suppressor protein of VHL). pVHL predominantly acts by the
direct regulation of the levels of hypoxia-inducible transcription
factor (HIF) types 1 and 2, which are significant in the cellular
response to oxygen deficiency. When VHL gene mutations (mostly
deletions) occur and pVHL is not produced, HIF is consequently not
blocked, resulting in the promotion of angioneogenesis and
uncontrolled cell proliferation. Thus, VHL gene mutations
significantly increase the risk of tumor growth in target organs
(8,9). The present study describes the case of
a 20-year-old male who suffered from obstructive jaundice due to
VHL disease-associated pancreatic lesions. The patient provided
written informed consent for the publication of this study.
Case report
In 1998, a 20-year-old male was admitted to the
General Hospital of Tianjin Medical University (Tianjin, China)
with symptoms of dizziness, unsteadiness and nausea for three
weeks, as well as vomiting for one week. Magnetic resonance imaging
(MRI) revealed a mass in the cerebellar vermis and surgery was
performed to excise the tumor. The pathological examination
identified a hemangioblastoma. Three years later, the patient was
readmitted to hospital due to dizziness lasting for one week. An
MRI examination showed recurrence of the hemangioblastoma in the
cerebellum and the patient underwent surgery to resect the tumor.
In April 2012, the patient was readmitted to hospital for the third
time due to jaundice. An enhanced computed tomography (CT) scan of
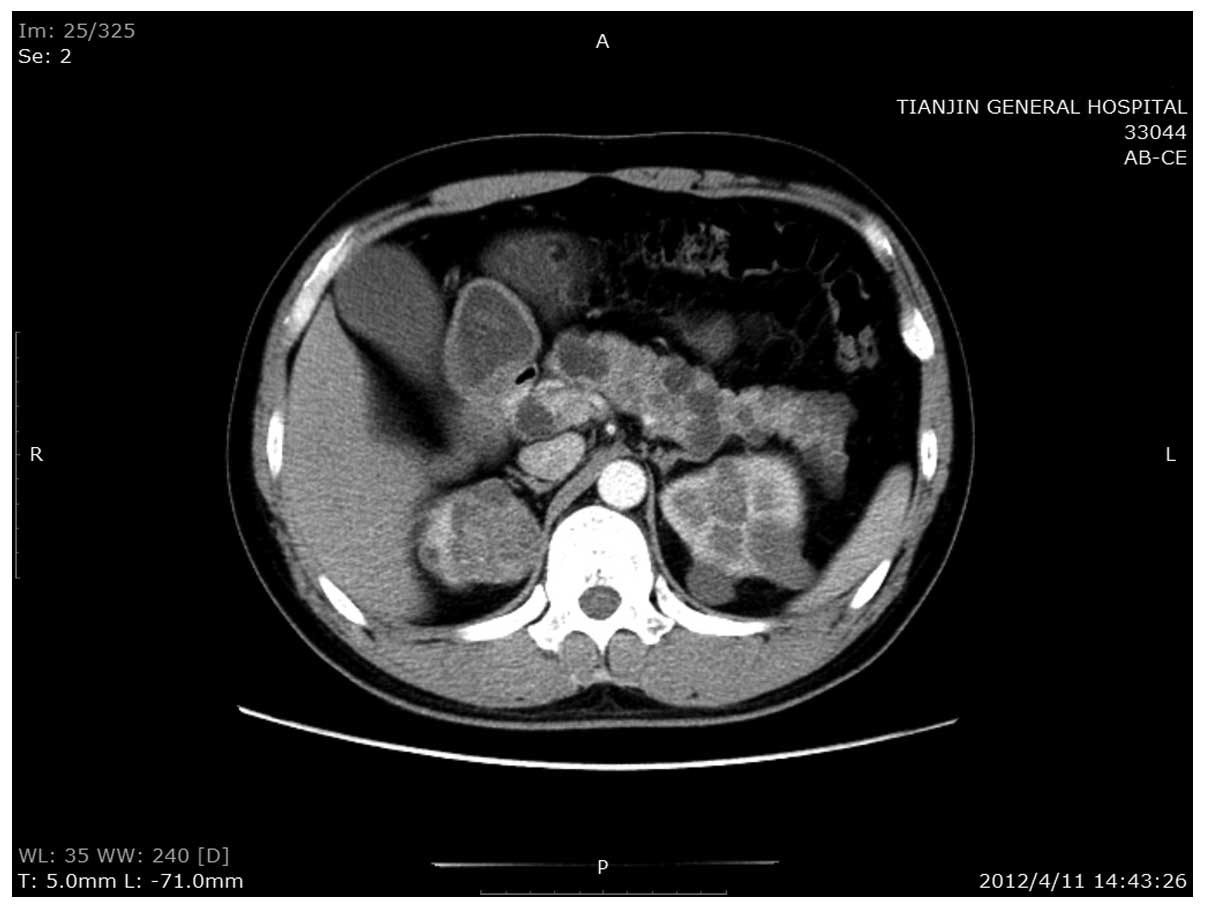
the abdomen revealed multiple RCCs in the kidneys, and a nodule
with a rich blood supply in the pancreatic head. In addition,
numerous cysts were identified throughout the pancreas (Fig. 1). These observations were confirmed
by magnetic resonance cholangiopancreatography, which revealed that
the nodule in the pancreatic head was ~2.9×2.2 cm in size and
possibly a NET. Due to the size and location of the tumor, the bile
duct in the pancreas was compressed and the upper parts of the
common bile and hepatic ducts were dilated. Since it was not
possible to excise the RCCs, the patient also refused surgery to
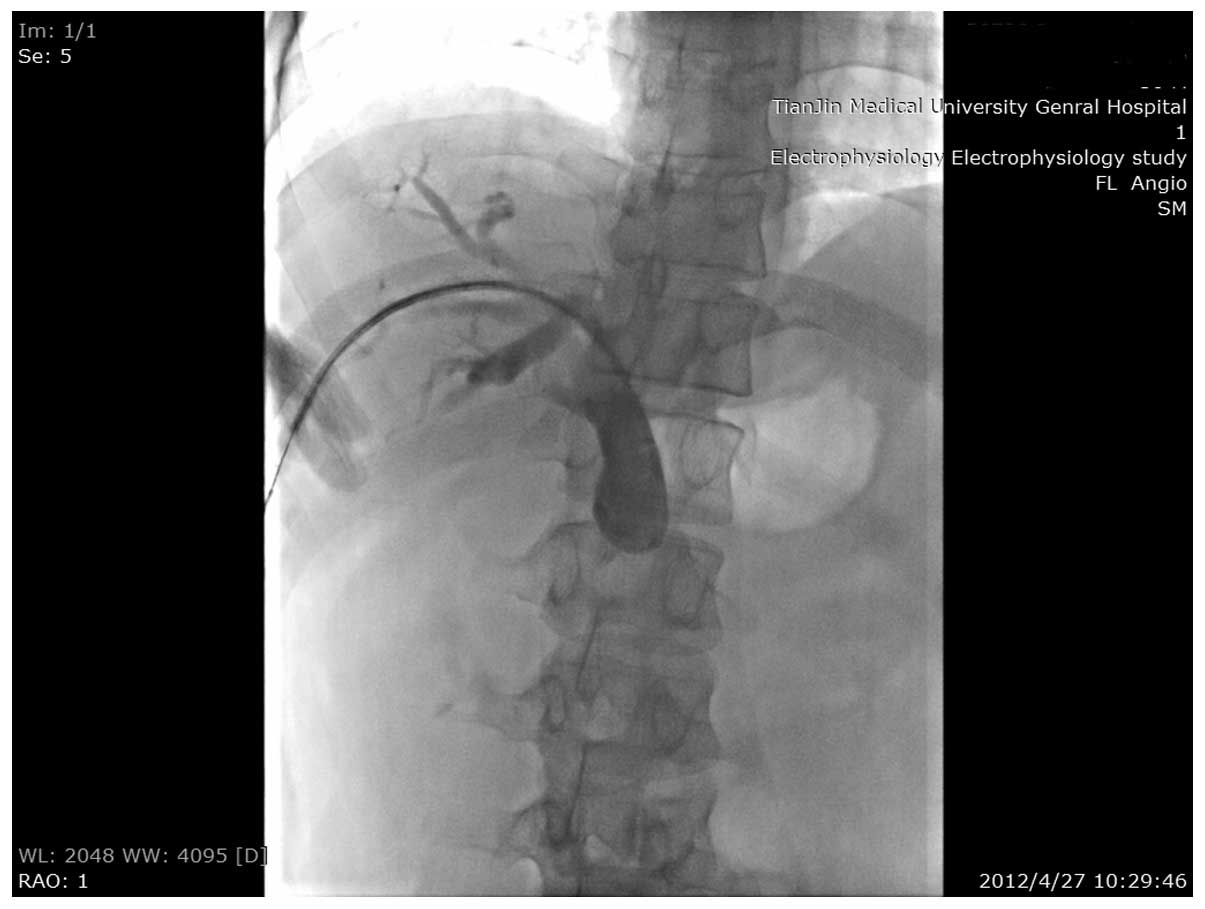
resect the pancreatic head mass. A metallic stent was placed at the
stenosis site of the common bile duct, which alleviated the
jaundice (Figs. 2 and 3). Nine months later, the patient returned
to the hospital with a fever, abdominal pain and jaundice. An
enhanced abdominal CT was performed, which revealed no change in
the size of the pancreatic head mass. The patient’s symptoms were
relieved following anti-inflammatory therapy for one week. However,
the patient continued to suffer the same symptoms every two months,
and gradually, anti-inflammatory therapy failed to alleviate the
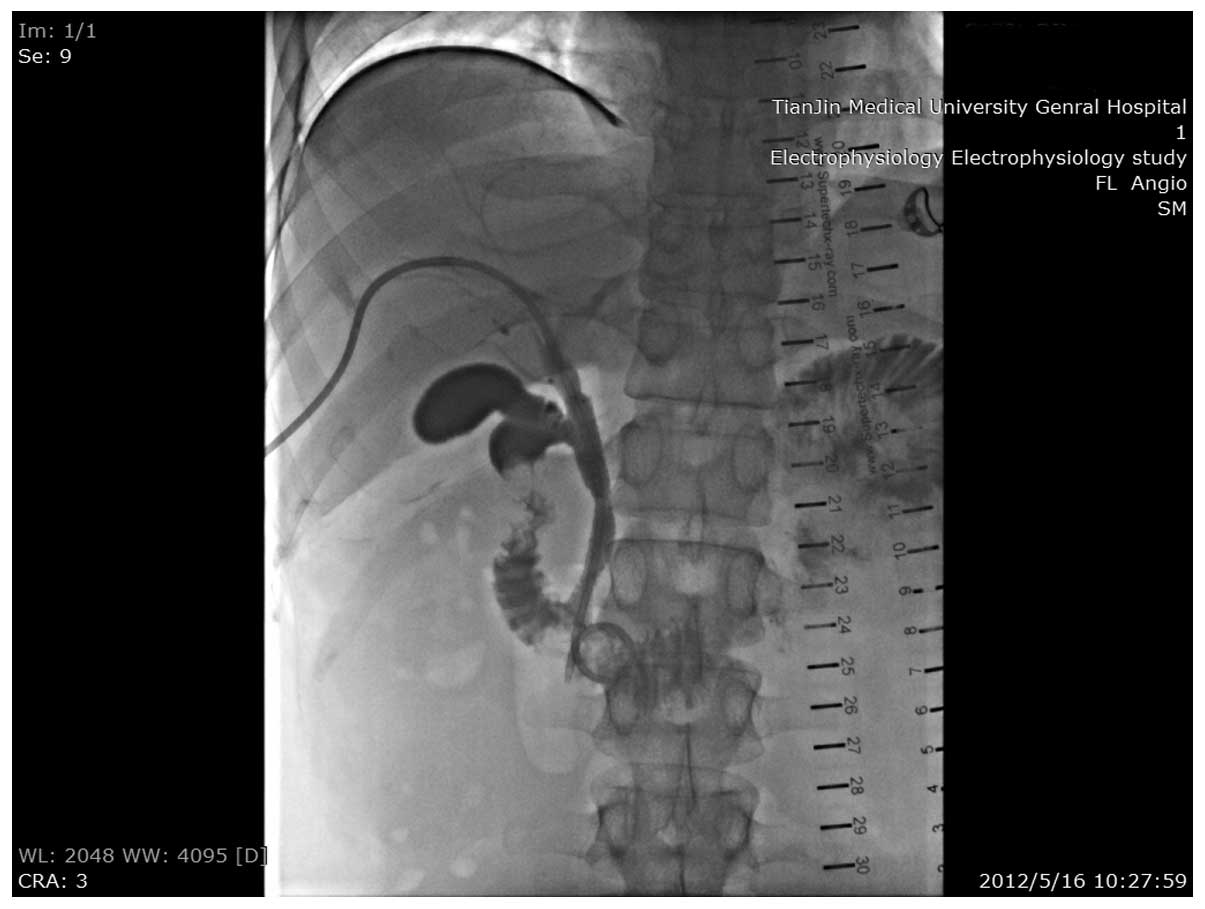
symptoms. Radiography tests revealed complete blockage of the stent
and thus, percutaneous transhepatic cholangiography (PTCD) surgery
was performed.
Genomic DNA was extracted from the peripheral blood
leukocytes and polymerase chain reaction was performed. Direct
sequencing revealed a known mutation of a base pair change at
nucleotide 473 in exon 3 (T473T/C) of the VHL gene, resulting in
the amino acid change Leu158Pro.
There was a recorded family history of VHL disease,
with the patient’s mother, grandmother, two uncles and three aunts
also suffering from the disease. Additionally, one uncle had
succumbed to RCC and five other individuals in the family had
succumbed to cerebral hemangioblastomas.
Discussion
VHL-associated pancreatic lesions are usually
non-functional and asymptomatic. A number of different pancreatic
lesions have been associated with VHL disease, including true
cysts, serous cystadenomas and NETs. In the majority of cases,
VHL-associated pancreatic NETs are also non-functional,
asymptomatic and typically slow growing (10). Charlesworth et al (11) reviewed 11 studies (excluding case
studies) of VHL-associated pancreatic lesions and identified that
211 (15%) of the 1,442 patients with VHL also exhibited pancreatic
NETs. Furthermore, metastasis was observed in 27 VHL patients
(12.8%) of the 211 patients with concurrent pancreatic NET, whereas
in patients with primary pancreatic NET, distant metastases were
reported at diagnosis in 64% of patients. Libutti et al
(12) reported that the median size
of lesions in the pancreas of patients exhibiting metastatic
disease was 5 cm, whereas in patients without metastatic disease,
the median size was 2 cm. In the present study, the pancreatic NET
was also non-functional, and no marked change in size was
identified after one year. However, since the tumor was located at
the head of pancreas, the bile duct in the pancreas was compressed
and the upper parts of the common bile and hepatic ducts were
dilated. As a result, the patient suffered from severe jaundice and
abdominal pain. Although VHL-associated pancreatic NETs may be
asymptomatic, they may cause certain obstructive symptoms by
blocking the common bile and pancreatic ducts.
In patients without VHL, pancreatic NETs should be
treated according to the degree of differentiation and malignancy.
Surgical resection may be recommended following the evaluation of
the functionality, disease stage and metastasis of the tumor
(13). However, in patients with
VHL-associated pancreatic NET, surgical treatment must be selected
carefully as this type of tumor is rarely the direct cause of
mortality (14). Blansfield et
al (14) reported that
pancreatic NETs were the cause of mortality in only 0.3% of
patients with VHL (633 patients in total) and in 1.9% of patients
exhibiting concurrent pancreatic NET and VHL (108 patients). In the
majority of cases, the cause of mortality in patients with VHL is
CNS hemangioblastoma or renal cancer. Accordingly, the prognosis of
pancreatic NET associated with VHL is considered to be favorable.
Therefore, in patients with VHL, the pancreatic NETs must be
treated based on tumor size. Libutti et al (12,15)
recommended surgery when the tumor size is ≥3 cm in the pancreatic
tail region and ≥2 cm in the pancreatic head region. Furthermore,
Blansfield et al (14)
recommended surgery when the tumor size is ≥3 cm. The studies also
proposed a tumor doubling time of <500 days as an additional
factor to be considered when selecting surgical treatment.
As the tumor in the present case was located at the
head of the pancreas and was >2 cm in size, according to the
standard reported by Libutti et al (15) surgery to resect the tumor was the
recommended treatment. However, the patient also suffered from
unresectable bilateral RCC, so whether it is reasonable to perform
a procedure, such as Whipple surgery, in such a case requires
further discussion. Based on the evidence that the RCC was
incurable, the patient refused Whipple surgery. The manner in which
to deal with such a case also requires investigation, as to date,
no studies have analyzed this problem. For the current patient, a
metallic stent was placed at the stenosis site of the common bile
duct, however, complete blockage of the stent was revealed nine
months later. PTCD was performed, but it is expected that the
effects of the surgery will not last long.
References
|
1
|
Richard S, Graff J, Lindau J and Resche F:
Von Hippel-Lindau disease. Lancet. 363:1231–1234. 2004.
|
|
2
|
Molino D, Sepe J, Anastasio P and De Santo
NG: The history of von Hippel-Lindau disease. J Nephrol. 19(Suppl
10): S119–S123. 2006.
|
|
3
|
Kim JJ, Rini BI and Hansel DE: Von Hippel
Lindau syndrome. Adv Exp Med Biol. 685:228–249. 2010.
|
|
4
|
Lonser RR, Glenn GM, Walther M, et al: von
Hippel-Lindau disease. Lancet. 361:2059–2067. 2003.
|
|
5
|
Tootee A and Hasani-Ranjbar S: Von
hippel-lindau disease: a new approach to an old problem. Int J
Endocrinol Metab. 10:619–624. 2012.
|
|
6
|
Maher ER, Neumann HP and Richard S: Von
Hippel-Lindau disease: a clinical and scientific review. Eur J Hum
Genet. 19:617–623. 2011.
|
|
7
|
Zbar B, Kishida T, Chen F, et al: Germline
mutations in the Von Hippel-Lindau disease (VHL) gene in families
from North America, Europe, and Japan. Hum Mutat. 8:348–357.
1996.
|
|
8
|
Clark PE and Cookson MS: The von
Hippel-Lindau gene: turning discovery into therapy. Cancer.
113(Suppl 7): S1768–S1778. 2008.
|
|
9
|
Baldewijns MM, van Vlodrop IJ, Vermeulen
PB, et al: VHL and HIF signalling in renal cell carcinogenesis. J
Pathol. 221:125–138. 2010.
|
|
10
|
Tamura K, Nishimori I, Ito T, Yamasaki I,
Igarashi H and Shuin T: Diagnosis and management of pancreatic
neuroendocrine tumor in von Hippel-Lindau disease. World J
Gastroenterol. 16:4515–4518. 2010.
|
|
11
|
Charlesworth M, Verbeke CS, Falk GA, et
al: Pancreatic lesions in von Hippel-Lindau disease? A systemic
review and meta-synthesis of the literature. J Gastrointest Surg.
16:1422–1428. 2012.
|
|
12
|
Libutti SK, Choyke PL, Alexander HR, et
al: Clinical and genetic analysis of patients with pancreatic
neuroendocrine tumors associated with von Hippel-Lindau disease.
Surgery. 128:1022–1028. 2000.
|
|
13
|
NCCN Clinical Practice Guidelines in
Oncology (NCCN Guidelines™). Neuroendocrine tumors. Version 1.
National Comphrensive Cancer Network; Fort Washington, PA: pp.
122011
|
|
14
|
Blansfield JA, Choyke L, Morita SY, Choyke
PL, Pingpank JF, Alexander HR, et al: Clinical, genetic and
radiographic analysis of 108 patients with von Hippel-Lindau
disease (VHL) manifested by pancreatic neuroendocrine neoplasms
(PNETs). Surgery. 142:814–818.e2. 2007.
|
|
15
|
Libutti SK, Choyke PL, Bartlett DL, Vargas
H, Walther M, Lubensky I, et al: Pancreatic neuroendocrine tumors
associated with von Hippel Lindau disease: diagnostic and
management recommendations. Surgery. 124:1153–1159. 1998.
|