Introduction
The occurrence of primary hepatic lymphoma (PHL) is
infrequent, and is responsible for <1% of all extranodal
lymphomas (1,2). The pathological diagnosis is usually
diffuse large B-cell lymphoma, and primary T-cell lymphoma of the
liver is extremely rare with only a few cases reported in the
literature (3), and is responsible
for 5–10% of PHLs (4). In the
present study, a case of primary hepatic peripheral T-cell lymphoma
in a middle-aged male patient is reported with a brief review of
the literature. Patient provided written informed consent.
Case report
Case presentation
A 59-year-old male patient presented to Zhongnan
Hospital of Wuhan University (Wuhan, China), on May 17, 2013, with
fatigue, weight loss of 20 kg and a three-day history of right
upper abdominal pain. The patient had no history of fever,
vomiting, night sweats, chest pain, icterus, diarrhea or stool
blood loss. The general physical and chest examinations of the
patient were unremarkable, except for right upper quadrant
tenderness, with no peripheral lymphadenopathy. The past medical
and personal histories of the patient were hypertension and
hyperlipidaemia for 5 years, and diabetes mellitus for 2 years.
Laboratory results included a hemoglobin level of 15.0 g/dl and a
white blood cell count of 8.3×109/l, with a normal
differential. Further laboratory investigation revealed an alanine
aminotransferase (ALT) level of 175 U/l and an aspartate
aminotransferase (AST) level of 222 U/l, and other liver and renal
function tests were within normal limits. Levels of serum
α-fetoprotein (AFP), carcinoembryonic antigen (CEA) and other tumor
markers were normal. Serology was negative for human
immunodeficiency (HIV), syphilis antibody, hepatitis C (HCV) and
hepatitis B (HBV) viruses. The patient had a serum lactate
dehydrogenase (LDH) level of 441 UI/ml (normal range, 135–225
UI/ml), and the level of β2-microglobulin was normal (1.38
mg/l).
Imaging
The chest X-ray did not show any abnormality.
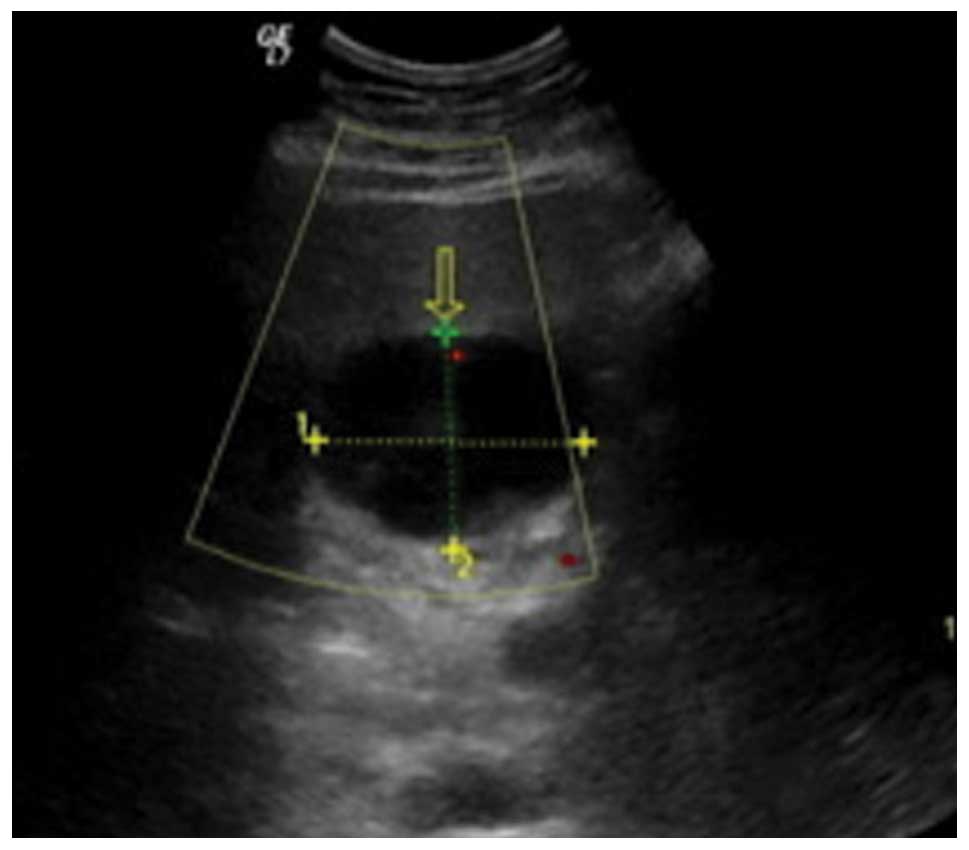
Abdominal ultrasonography (US) showed a well-defined hypoechoic
mass of 53×39 mm in the quadrate lobe of the liver, and the
internal echo was heterogeneous (Fig.
1). Diagnostic imaging was performed by computed tomography
(CT) and a magnetic resonance imaging (MRI) scan of the abdomen. On
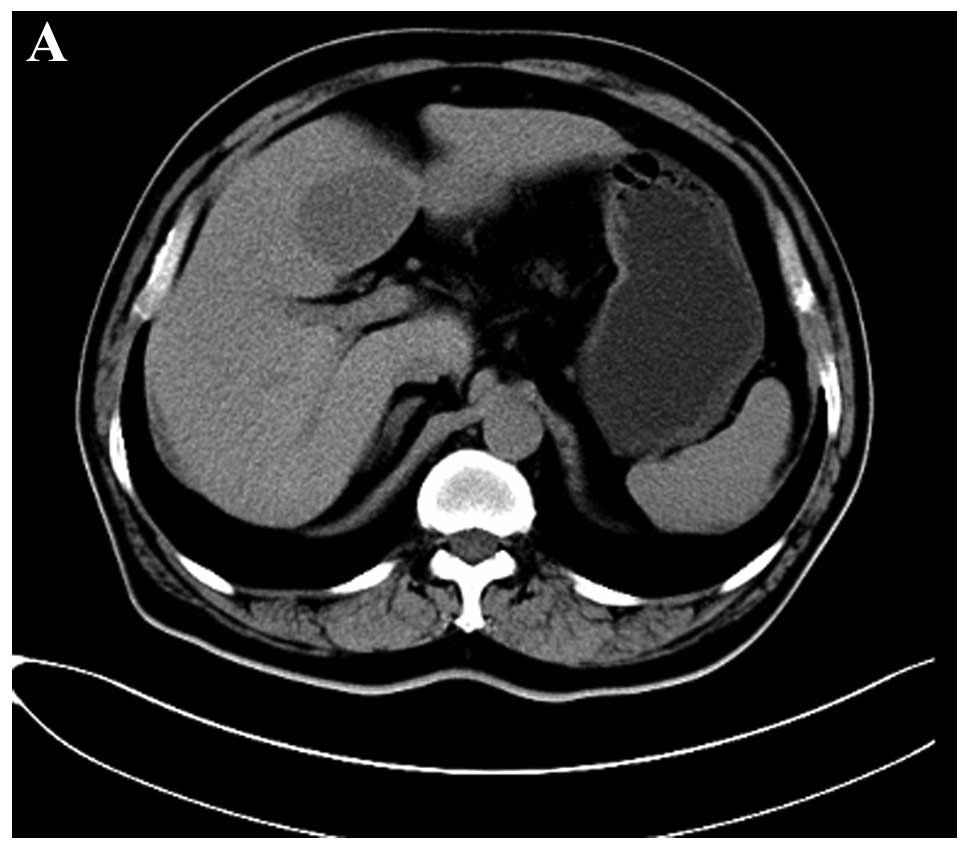
abdominal CT scan (Siemens Somatom Definition; Siemens Medical
Solutions, Erlangen, Germany), an oval homogenous and low-density
mass that measured ~40×58 mm in the largest section with a distinct
border located in the quadrate lobe of liver was demonstrated prior
to contrast material injection (Fig.
2A). On triple-phase (arterial, portal venous and delayed
phase) iodinated contrast-enhanced CT scan, a slight and persistent
ring-like enhancement was visible in the peripheral but not in the
entire tumor, the center of which was minimally enhanced (Fig. 2B–D). Supplemental abdominal MRI with
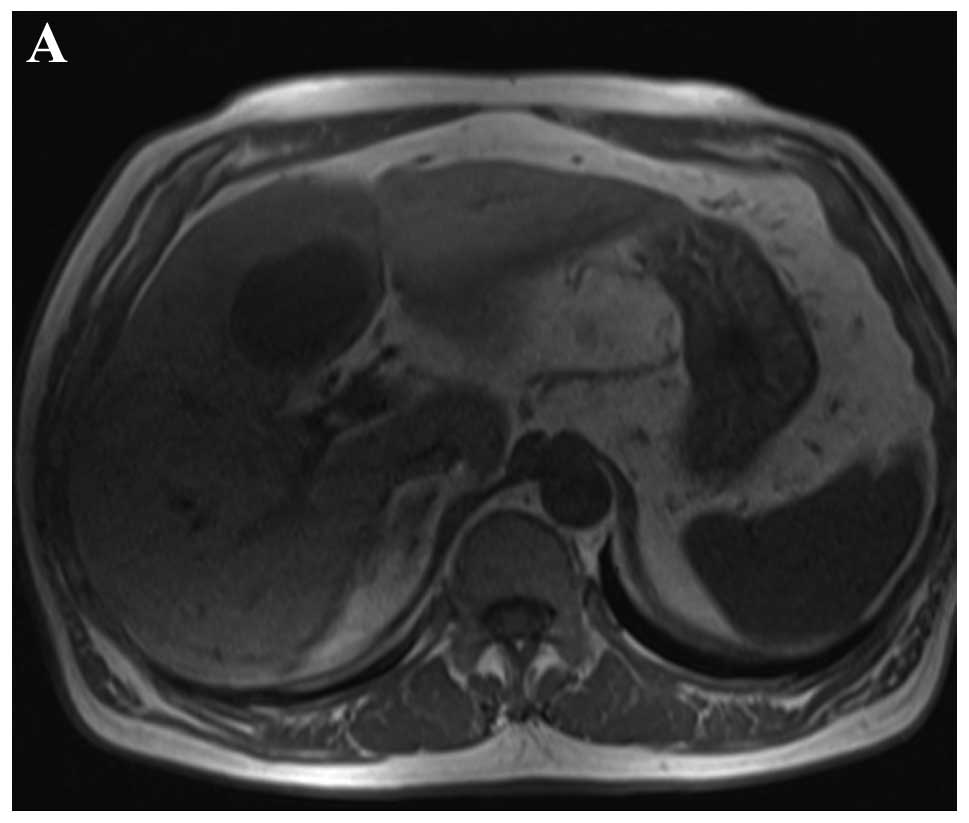
contrast medium (Siemens Trio 3.0T; Siemens Medical Solutions)
showed a homogeneous and distinct solitary lesion at the fourth
hepatic segment, which had a low signal intensity on T1 weighted
image (WI) and a high signal intensity on T2WI (Fig. 3A and B). The dynamic
gadolinium-diethylenetriaminepentaacetic acid MRI protocol showed a
mild ring-like enhancement during the arterial phase, which
continued and showed a prominent enhancement in the portal venous
phase. The enhancement of the tumor decreased in the delayed phase
and showed the enhancement of the septum (Fig. 3C–E).
Surgery and pathological analysis
The patient underwent total resection of the mass.
Preoperatively, the mass measured 60×40 mm and it was lobulated,
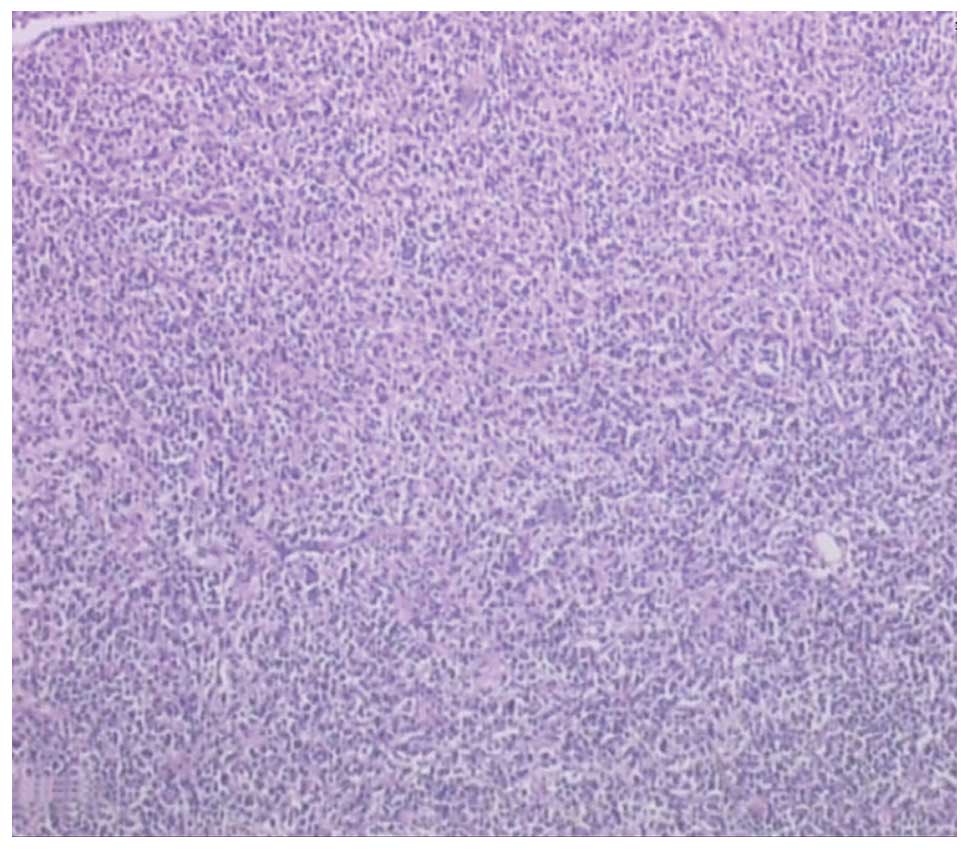
well-defined and had necrosis at the centre. Histopathological
analysis of the tissue disclosed a diffuse infiltrate with
medium-to-large-sized lymphoid cells indicative of lymphoma
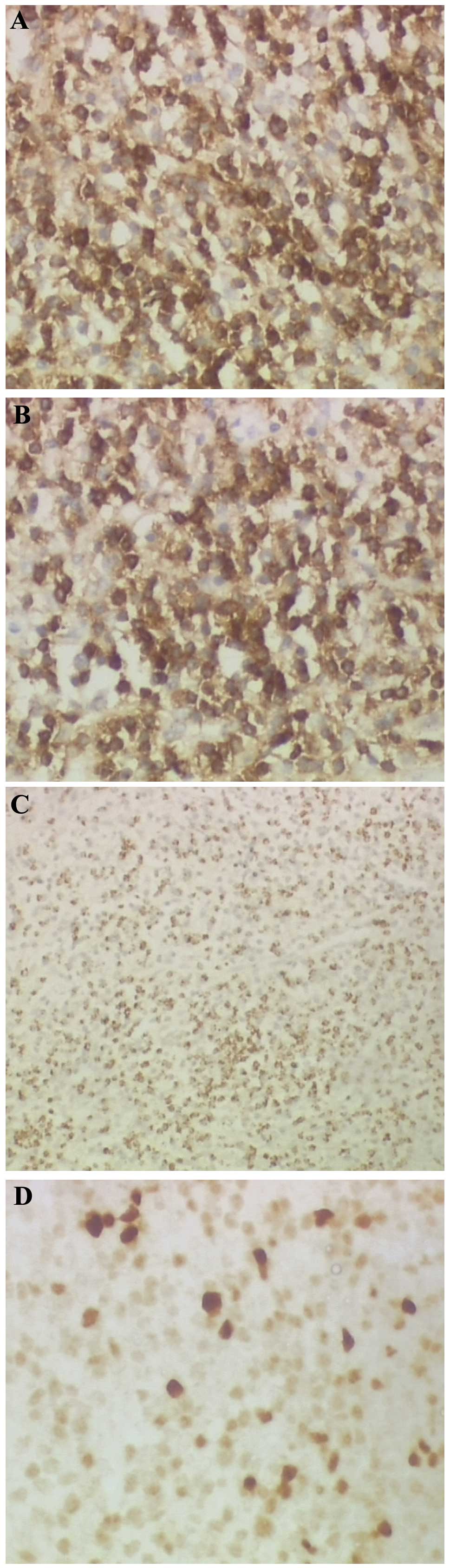
(Fig. 4). Immunohistochemistry of
the tumor cells showed reactivity for cluster of differentiation 3
(CD3) (Fig. 5A), CD5 (Fig. 5B), TIA-1 (Fig. 5C) and multiple myeloma oncogene 1
(Fig. 5D), and was negative for
CD20, CD79, activin receptor-like kinase-1, CD30, CD10,
myeloperoxidase, B-cell lymphoma 6 and smooth muscle actin. The
Ki-67 index of those lymphoid cells was 30%.
Chemotherapy and follow-up
Following a discussion of the risks of chemotherapy
and radiotherapy with the patient and his family, the patient
received chemotherapy (CHOP: 1500 mg cyclophosphamide, 150 mg
epirubicin-adriamycin, 2 mg vincristine and 100 mg prednisone). The
courses of chemotherapy were administered every 21 days. Subsequent
to receiving six cycles of chemotherapy, the patient underwent
radiotherapy of liver (Dt = 30Gy/15F). During the treatment period
with chemotherapy and radiotherapy, there were no major
complications. The patient has undergone follow-up for almost 1
year with no evidence for recurrence of the disease.
Discussion
According to the criteria by Caccamo et al,
PHL is established as being a lymphoma with only the involvement of
the liver at presentation. Six months after the diagnosis, other
tissues can be involved, including the spleen, lymph nodes,
peripheral blood, bone marrow or other tissues (5). PHL is notably rare, it constitutes
0.4% of cases of extranodal non-Hodgkin’s lymphoma (NHL), and only
~0.016% of all cases of NHL (6).
The most common histological type of PHL is diffuse large B-cell
lymphoma, and primary hepatic T-cell lymphoma is extremely rare
with only a few cases reported in the literature, which are
responsible for 5–10% of PHLs (3).
The etiology of PHL is unknown, despite certain
possible etiological factors having been proposed, including HCV
(7–9), HBV (10) and Epstein-Barr virus (EBV) (11). HCV infection has been identified in
20–60% of PHL patients. The persistent correlation with HCV
indicates that this virus may play a role in PHL pathogenesis
(7,12). PHL has been noted to occur in
patients with immune suppression, such as HIV or human
T-lymphotropic virus infections, systemic erythematous lupus and
immunosuppressive therapy (2,11).
However, the patient of the present study had neither HCV infection
nor signs of immunodeficiency, due to negative serology for HIV,
HBV, HCV and EBV active infection. Therefore, it is speculated that
PHL could also occur in patients without any liver disease.
PHL commonly occurs at 50–60 years of age, with a
male/female ratio of 2–3/1 (13).
PHL has non-specific clinical manifestations. The most frequent
symptom at presentation is abdominal pain or discomfort, occurring
in 39–70% of patients (3), and
other symptoms include fever, loss of weight and night sweats (also
known as ‘B’ symptoms), nausea, vomiting, asthenia or itching. The
main laboratory findings are abnormal hepatic functional enzymes,
including AST, ALT, bilirubin, γ-glutamyl transferase, ALP, and
LDH. Liver function tests are abnormal in <70% of cases and LDH
is elevated in 30–80% of patients (2,11).
Another study has also revealed that the dynamic change of serum
LDH could serve as a diagnostic marker (14), but its use is limited due to poor
specificity. β2-microglobulin, a well-described prognostic marker
in lymphoma, is elevated in >90% of patients (12). AFP and CEA are tumor markers that
are present at normal levels in ~100% of patients with PHL, which
assists the differential diagnosis (12,15).
In the present case, the levels of serum LDH, ALT and AST were
elevated, those of AFP and CEA were normal, and the level of
β2-microglobulin was normal.
At presentation, PHL may be a solitary lesion,
multiple lesions or it may diffuse infiltration of the liver
(16). The most common
manifestation is a solitary lesion, and the diffuse infiltration is
rare and indicates a worse prognosis. The imaging appearance of
hepatic lymphoma is non-specific and, on ultrasound, the lesions
usually appear hypoechoic with no typical vascularization pattern
(3,17). PHL lesions appear as hypoattenuating
in CT scans, which may have a low-intensity central area with no
enhancement following the administration of an intravenous contrast
in half the cases, patchy enhancement in 33% of patients and a ring
of enhancement in ~25% of cases (3,17,18).
Classically, MRI findings in PHL are described as ‘hypointense’ or
‘isointense’ on T1WI, and ‘hyperintense’ on T2WI (3,19). The
imaging findings of hepatic lymphoma in the present case were
similar to previous studies. Pre-contrast CT and MRI scans revealed
that the mass was homogeneous and had a well-defined margin.
Contrast-enhanced CT and MRI showed a ring-like enhancement.
Due to the rarity of this disease, non-specific
clinical symptoms and laboratory and radiological manifestations,
the diagnosis of PHL is extremely difficult. PHL may be confused
with other diseases, including primary hepatic carcinoma,
metastases and focal nodular hyperplasia. Laboratory and imaging
findings are extremely helpful in differentiating between PHL and
these diseases. Primary hepatic carcinoma appears as hyperechoic
lesion in ultrasound, and CT scans show prominent arterial
enhancement and iso- or hypodense on portal venous and delayed
phases. The level of AFP is often elevated and the hepatic
metastases have the history of a primary tumor generally. Focal
nodular hyperplasia (FNH) usually appears hypo- or isodense on CT,
and isointense on MRI. FNH is fairly homogeneous except for the
central scar, which typically is hypodense on CT and T2-bright on
MRI. The central scar is extremely specific. FNH shows rapid uptake
of contrast in the arterial phase with a rapid return to
near-normal enhancement in the portal venous and delayed phases.
The central scar may enhance slightly in the delayed phase
(20). However, PHL presents
hypoechoic in US and low density in the CT scan. PHL shows no
enhancement, minimally patchy or ring-like enhancement in
contrast-enhanced CT, and delayed enhancement in the portal venous
and delayed phases. The level of AFP and other tumor markers are
normal. As the patient of the present study showed, the lesion was
hypoechoic on ultrasound and low-density, minimally ring-enhancing
on CT scan. For the MRI scan, the lesion presented a low signal
intensity on T1WI and high signal intensity on T2WI. Combining the
clinical and laboratory features, the diagnosis of PHL can be
speculated. However, the definite diagnosis requires histological
results by liver biopsy or surgical resection and the absence of
lymphoproliferative disease outside the liver. The patient in the
present case underwent surgical resection, and liver biopsy stained
with specific immunohistochemical stains confirmed the diagnosis of
PHL.
The optimal treatment of PHL has not yet been
defined, however surgical treatment, radiotherapy and chemotherapy
have been reported as treatment modalities both alone and in
combination (21). It has been
reported that surgical resection alone or in combination with
chemotherapy may be a good treatment option for low-volume
localized PHL (3,22). The patient of the present study
employed surgical treatment and subsequent chemotherapy and
radiotherapy in combination.
The majority of patients with PHL present with a
poor prognosis. The median survival time for all patients is 15.3
months; however, the variation is wide and the reported survival
time ranges from 3 to 123.6 months (11). In specific reports, the prognosis
has been linked to the pattern of liver involvement (23) and the pathological subtype (3), and it is known that patients with
unfavorable histologies have a low survival rate. The study by
Emile et al (23) observed
that in patients with nodular hepatic involvement, 1- and 3-year
survival rates were 70 and 57%, respectively; however, when the
liver was diffusely involved, the 1- and 3-year survival rates
dropped to 38 and 18%, respectively. Therefore, it can be deduced
that the patients with nodular involvement of the liver will have a
longer survival rate. Yang et al (24) revealed that postoperative
chemotherapy was the only significant prognostic factor that
influenced survival rate. Noronha et al (3) reported that a patient, who was alive 5
years following the initial diagnosis, was treated with surgery
followed by chemotherapy and radiation. A study by Lei (2) indicated that adjuvant chemotherapy
subsequent to surgery should be considered for treatment of
patients with localized disease to prevent recurrence. Therefore,
we believe that a good prognosis can be achieved by early surgery
combined with chemotherapy in patients with localized disease (such
as solitary nodular PHL) and favorable histology
In conclusion, PHL is a notably infrequent disease,
which lacks established imaging, clinical and biochemical markers.
The diagnosis is difficult, as it is impossible to differentiate a
single non-Hodgkin hepatic lesion from a metastatic nodule only by
imaging techniques, particularly in the case of a history of tumor
in a patient with unremarkable physical examination and no B
symptoms. Biopsy or surgical resection should be performed when
possible in case of an isolated hepatic nodule with radiological
malignant aspects, particularly when serum tumor markers or
biochemistry are not informative, as only histology can ensure an
accurate differential diagnosis.
Acknowledgements
The authors would like to thank Zhi-Gao Xu for
obtaining the pathological and immunohistochemical photomicrographs
presented.
References
|
1
|
Harris AC, Ben-Ezra JM, Contos MJ and
Kornstein MJ: Malignant lymphoma can present as hepatobiliary
disease. Cancer. 78:2011–2019. 1996.
|
|
2
|
Lei KI: Primary non-Hodgkin’s lymphoma of
the liver. Leuk Lymphoma. 29:293–299. 1998.
|
|
3
|
Noronha V, Shafi NQ, Obando JA and Kummar
S: Primary non-Hodgkin’s lymphoma of the liver. Crit Rev Oncol
Hematol. 53:199–207. 2005.
|
|
4
|
Salmon JS, Thompson MA, Arildsen RC and
Greer JP: Non-Hodgkin’s lymphoma involving the liver: clinical and
therapeutic considerations. Clin Lymphoma Myeloma. 6:273–280.
2006.
|
|
5
|
Caccamo D, Pervez NK and Marchevsky A:
Primary lymphoma of the liver in the acquired immunodeficiency
syndrome. Arch Pathol Lab Med. 110:553–555. 1986.
|
|
6
|
Freeman C, Berg JW and Cutler SJ:
Occurrence and prognosis of extranodal lymphoma. Cancer.
29:252–260. 1972.
|
|
7
|
Bronowicki JP, Bineau C, Feugier P, et al:
Primary lymphoma of the liver: clinical-pathological features and
relationship with HCV infection in French patients. Hepatology.
37:781–787. 2003.
|
|
8
|
Kuroda J, Omoto A, Fujiki H, et al:
Primary hepatic Burkitt’s lymphoma with chronic hepatitis C. Acta
Haematol. 105:237–240. 2001.
|
|
9
|
Yago K, Shimada H, Itoh M, et al: Primary
low-grade B-cell lymphoma of mucosa-associated lymphoid tissue
(MALT)-type of the liver in a patient with hepatitis C virus
infection. Leuk Lymphoma. 43:1497–1500. 2002.
|
|
10
|
Aozasa K, Mishima K and Ohsawa M: Primary
malignant lymphoma of the liver. Leuk Lymphoma. 10:353–357.
1993.
|
|
11
|
Avlonitis VS and Linos D: Primary hepatic
lymphoma: a review. Eur J Surg. 165:725–729. 1999.
|
|
12
|
Page RD, Romanguera JE, Osborne B, et al:
Primary hepatic lymphoma: favorable outcome after combination
chemotherapy. Cancer. 92:2023–2029. 2001.
|
|
13
|
Haider FS, Smith R and Khan S: Primary
hepatic lymphoma presenting as fulminant hepatic failure with
hyperferritinemia: a case report. J Med Case Rep. 2:2792008.
|
|
14
|
Taketomi A, Takenaka K, Shirabe K, et al:
Surgically resected primary malignant lymphoma of the liver.
Hepatogastroenterology. 43:651–657. 1996.
|
|
15
|
Lei KI, Chow JH and Johnson PJ: Aggressive
primary hepatic lymphoma in Chinese patients. Presentation,
pathologic features and outcome. Cancer. 76:1336–1343. 1995.
|
|
16
|
Levy AD: Malignant liver tumors. Clin
Liver Dis. 6:147–164. 2002.
|
|
17
|
Elsayes KM, Menias CO, Willatt JM, Pandya
A, Wiggins M and Platt J: Primary hepatic lymphoma: imaging
findings. J Med Imaging Radiat Oncol. 53:373–379. 2009.
|
|
18
|
Maher MM, McDermott SR, Fenlon HM, Conroy
D, O’Keane JC, Carney DN and Stack JP: Imaging of primary
non-Hodgkin’s lymphoma of the liver. Clin Radiol. 56:295–301.
2001.
|
|
19
|
Kelekis NL, Semelka RC, Siegelman ES, et
al: Focal hepatic lymphoma: magnetic resonance demonstration using
current techniques including gadolinium enhancement. Magn Reson
Imaging. 15:625–636. 1997.
|
|
20
|
Terkivatan T, van den Bos IC, Hussain SM,
Wielopolski PA, de Man RA and IJzermans JN: Focal nodular
hyperplasia: lesion characteristics on state-of-the-art MRI
including dynamic gadolinium-enhanced and superparamagnetic
iron-oxide-uptake sequences in a prospective study. J Magn Reson
Imaging. 24:864–872. 2006.
|
|
21
|
Agmon-Levin N, Berger I, Shtalrid M,
Schlanger H and Sthoeger ZM: Primary hepatic lymphoma: a case
report and review of the literature. Age Ageing. 33:637–640.
2004.
|
|
22
|
Scoazec JY, Degott C, Brousse N, Barge J,
Molas G, Potet F and Benhamou JP: Non-Hodgkin’s lymphoma presenting
as a primary tumor of the liver: presentation, diagnosis and
outcome in eight patients. Hepatology. 13:870–875. 1991.
|
|
23
|
Emile JF, Azoulay D, Gornet JM, et al:
Primary non-Hodgkin’s lymphomas of the liver with nodular and
diffuse infiltration patterns have different prognoses. Ann Oncol.
12:1005–1010. 2001.
|
|
24
|
Yang XW, Tan WF, Yu WL, et al: Diagnosis
and surgical treatment of primary hepatic lymphoma. World J
Gastroenterol. 16:6016–6019. 2010.
|