Introduction
Glioblastomas (GBMs) are tumors that arise from
astrocytes, featuring high metastasis, recurrence and mortality
rates. Angiogenesis plays an essential role in the growth,
proliferation, migration, invasion and metastasis of GBM. Targeting
angiogenesis is an important way to treat GBM at present. The
angiogenic vascular endothelial growth factor (VEGF)/VEGF receptor
(VEGFR) pathway has been considered the most important signaling
pathway of GBM angiogenesis (1).
However, bevacizumab, a humanized monoclonal antibody directed
against VEGF-A, which has recently received regulatory approval for
the treatment of recurrent GBM, has limited clinical responses
(2,3). Future targeted angiogenesis therapy
with effective drugs will provide novel avenues for the treatment
of malignant gliomas.
Vasculogenic mimicry (VM) is known as the formation
of non-endothelial tumor cell-lined microvascular channels in
aggressive tumors, and does not involve endothelial cells. VM is
detected predominantly in high-grade gliomas and there is
significant association between VM and glioma grade. VM has also
been demonstrated to be involved in the proliferation, invasion and
metastasis of malignant glioma (4–6). The
existence of VM may provide compensation to ensure a blood supply,
particularly in less vascularized regions of the tumor. VM may be a
novel therapeutic target for anti-angiogenesis in GBM. The
molecular mechanisms through which GBM forms VM remain elusive.
Previous studies on melanoma have indicated that
erythropoietin-producing hepatocellular carcinoma-A2 (EphA2),
phosphoinositide 3-kinase (PI3K) and matrix metalloproteinases
(MMPs) are the key factors for VM formation (7–11). In
melanoma VM, EphA2 is phosphorylated through interactions with its
membrane bound ligand, Ephrin-A1. Subsequently, phosphorylated
EphA2 activates PI3K, leading to enhanced MMP-14 and -2 expression.
MMP-14 and -2 can promote the cleavage of the laminin-5γ2 chain
into promigratory γ2′ and γ2× fragments. Fragment release into the
tumor microenvironment increases tumor cell migration and invasion,
and may ultimately result in VM formation. In malignant glioma VM,
it remains unknown whether there is a similar molecular mechanism
(10,11).
Curcumin (CCM), a naturally polyphenol derived from
the root of the rhizome Curcuma longa, possesses
anti-inflammatory, antioxidant and anticancer properties. CCM is
capable of suppressing carcinogenesis in the forestomach, skin,
mammary glands, liver and colon in various animal models in
vivo (12). Previous studies
had indicated that CCM suppresses angiogenesis in malignant tumors
(13–15). The anti-angiogenesis mechanism of
CCM is involved in various signal pathways, including the
downregulated expression of PI3K and MMP-2. As aforementioned, PI3K
and MMP-2 have previously been associated with VM formation. We
hypothesize that CCM may be able to suppress VM formation.
Recently, Chen et al reported that CCM inhibited tumor
growth and reduced VM in a murine choroidal melanoma model
(16). Tumor volume was reduced by
CCM and coincidently, the numbers of VM were also significantly
decreased. The expression levels of EphA2, PI3K and MMP-2 and -9
were also lower in the treatment group compared with the control
group. These observations suggested that CCM had the ability to
inhibit the growth of engrafted melanoma VM channels through the
regulation of vasculogenic factors that could be associated with
the downregulation of the EphA2/PI3K/MMPs signaling pathway. Based
on these previous studies, we hypothesized that CCM may affect VM
formation in GBM through the regulation of EphA2, PI3K and MMP-2
expression. To test this hypothesis, the present study first
analyzed the efficacy of CCM in affecting VM formation in malignant
GBM U251 cells, and then examined the expression of EphA2, PI3K and
MMP-2 by quantitative PCR and western blot assay following CCM
treatment.
Materials and methods
Cell culture and experimental groups
The human glioma U251 cell line was obtained from
the American Type Culture Collection (Manassas, VA, USA). CCM
(C21H20O6; >98% purity) was
purchased from Sigma Chemical Co. (St. Louis, MO, USA) and
dissolved in dimethyl sulfoxide (DMSO) to create a stock solution,
and stored at −20°C until use. The human malignant GBM U251 cell
line was maintained in RPMI-1640 cell culture medium supplemented
with 10% fetal bovine serum and 100 U/ml penicillin/streptomycin
(all Hyclone, GE Healthcare Life Sciences, Logan, UT, USA) in a
humidified incubator containing 5% CO2 at 37°C. Cultures
were passaged every 2–3 days after reaching 80% confluence. Cells
in the logarithmic growth phase were used in the experiment.
The cells were divided into the control group
(complete culture), the DMSO group (DMSO only, as the negative
control group) and the CCM group (treated with CCM at various
concentrations, as the treatment group).
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay for cell proliferation
The U251 cells were plated at 1×104 cells
per well in 96-well culture plates, incubated for 24 h. In
accordance with the experimental groups, various concentrations of
CCM (2.5, 5, 10, 20, 40 and 80 μM) were added to each well.
Subsequent to 12, 24, 48 and 72 h, MTS assays were performed
(CellTiter 96® Aqueous One Solution Cell Proliferation
Assay, Promega, Madison, WI, USA). MTS was added to each well at a
proportion of 1/10 and incubated for 4 h at 37°C. The medium was
then removed and 180 μl DMSO was added to each well. The absorbance
at 490 nm was measured by a microplate reader (multiscan MK3;
Thermo Fisher Scientific, Waltham, MA, USA). The mean cell
proliferation was calculated from the absorbance units. All the
experiments were carried out in triplicate and each experiment was
repeated three times.
Transwell migration assay
U251 cells at 1×105 per well were seeded
into the upper compartment of a Transwell Boyden chamber (BD
Biosciences, Franklin Lakes, NJ, USA), with 100 μl serum-free media
added into the upper compartment and 600 μl complete media added
into the lower compartment. In accordance with the experimental
groups, various concentrations of CCM (5, 10, 20 and 40 μM) were
added into the upper compartment. Subsequent to incubation for 48 h
at 37°C, images of the cells of each group that had migrated to the
chamber of the poly carbon membrane were captured and the results
quantified.
Transwell invasion assay
The Matrigel basement membrane matrix (BD
Bioscience) was mixed with serum-free medium at a proportion of
1:3, then applied to the upper compartment of the Transwell Boyden
chamber, 40 μl per well. Following 30 min of incubation at 37°C,
the Matrigel solidified. U251 cells at 1×105 per well
were seeded into the upper compartment, 100 μl serum-free media was
added into the upper compartment and 600 μl complete media was
added into the lower compartment. In accordance with the
experimental groups, various concentrations of CCM (5, 10, 20 and
40 μM) were added into the upper compartment. Following incubation
for 48 h at 37°C, images of the cells of each group that had
migrated to the chamber of the poly carbon membrane were captured
and the results quantified.
In vitro VM tube formation assay
The VM network was established as described
previously by Ling et al (17). Briefly, the Matrigel basement
membrane matrix (BD Biosciences) was thawed at 4°C, mixed quickly
with fetal bovine serum at a proportion of 1:3 and then 20 μl was
added to each of the 96-well plates. This was allowed to solidify
for 2 h at 37°C. The cells were adjusted to 1×105/ml and
seeded on the Matrigel-coated plates in a final volume of 200 μl
(2×104 cells per well). CCM at various concentrations
(5, 10, 20 and 40 μM) was added to each well. The cells were then
incubated in a humidified 5% CO2 incubator at 37°C.
Next, 24 h later, images of each well were captured using
phase-contrast microscopy and then quantified.
Quantitative PCR assay for measurement of
the mRNA expression of EphA2, PI3K and MMP-2
To evaluate the mRNA expression of EphA2, PI3K and
MMP-2 following CCM treatment, quantitative PCR was performed.
Briefly, the U251 cells plated at 1×104 per well in the
96-well culture plates were incubated with various concentrations
of CCM (5, 10, 20 and 40 μM) for 48 h. Total RNA from each sample
was then isolated with TRIzol reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA), according to the manufacturer’s
instructions. An optical density assay (RNA 260/280 ratio
determined) and agarose electrophoresis analysis showed that the
purity and integrity of the RNA was good. Total RNA (1 μg) was used
for the reverse transcription process with a Takara PrimeScript™ RT
Reagent kit with gDNA Eraser (Takara, Dalian, China), according to
the manufacturer’s instruction, and the first-strand cDNA was
prepared. The quantitative PCR was performed with Takara
SYBR® Premix Ex Taq™ II (Takara) on an ABI
PRISM® 7500 Sequence Detection System with a 10-μl total
reaction volume consisting of the following: 5 μl 2X SYBR premix EX
Taq™, 0.5 μl (10 μM) PCR forward primer, 0.5 μl (10 μM) PCR reverse
primer, 0.5 μl cDNA and 3.5 μl dH2O. The reaction
process was performed at 50°C for 2 min, 95°C for 2 min, 95°C for
15 sec and 60°C for 32 sec, for 40 cycles. The melting curve was
analyzed at the temperature range of 60°C–95°C. GAPDH served as an
internal reference. The primers were as follows: EphA2 forward,
5′-AAGACCCTGGCTGACTTT-′3 and reverse, 5′-GTTCACCTGGTCCTTGAGT-′3;
PI3K forward, 5′-AAAGGCGGCTTGAAAGGT-′3 and reverse,
5′-GACGATCTCCAATTCCCAAA-′3; MMP-2 forward,
5′-CTGGAGATACAATGAGGTGAAG-′3 and reverse,
5′-TCTGAGGGTTGGTGGGATTG-′3; and 18S rRNA forward,
5′-CCTGGATACCGCAGCTAGGA-′3 and reverse,
5′-GCGGCGCAATACGAATGCCCC-′3.
Western blot assay for measurement of the
protein expression of EphA2, PI3K and MMP-2
To evaluate the protein expression of EphA2, PI3K
and MMP-2 following CCM treatment, a western blot assay was
performed. Briefly, the U251 cells plated at 1×104 per
well in 96-well culture plates were incubated with various
concentrations of CCM (5, 10, 20 and 40 μM) for 48 h. Protein from
each sample was extracted with radioimmunoprecipitation assay cell
lysate (CST; Cell Signaling Technology, Inc., Danvers, MA, USA).
Protein quantitative analysis was evaluated with a bicinchoninc
acid protein assay kit (Pierce Biotechnology, Inc., Rockford, IL,
USA). The protein was heated with 5X SDS sample buffer, and 30 μl
sample/hole was separated by SDS-PAGE gel electrophoresis, then
transferred to polyvinylidene difluoride membranes and washed three
times in phosphate-buffered saline with Tween-20 (PBST), for 5 min
each time. The membranes were blocked with 5% bovine serum albumin
for 1 h at room temperature, and then incubated with the following
primary antibodies: Anti-EphA2 (1:500; rabbit polyclonal to Eph
receptor A2), anti-PI3K [1:1,000; mouse monoclonal (M253) to PI3K
p85-C-termina], anti-MMP-2 [1:400; mouse monoclonal
(CA-4001/CA719E3C) to MMP-2) (all Abcam, Cambridge, UK), incubated
overnight at 4°C. Subsequent to being thoroughly washed with TBST
three times, horseradish peroxidase (HRP)-conjugated secondary
antibodies (goat anti-mouse immunoglobulin G-HRP; Wuhan Boster
Biological Technology, Ltd., Wuhan, China) were applied, incubated
for 2 h at room temperature and washed three times in PBST.
Enhanced chemiluminescence light-emitting liquid was then added.
GAPDH-HRP (KangChen Bio-tech, Inc., Shanghai, China) was used as an
internal reference. The gray scale value of each sample was
detected with the fluorescence imaging analyzer (BioPhotometer
plus; Eppendorf, Hamburg, Germany).
Statistical analysis
All experiments were performed three times. The data
were recorded as the mean ± standard deviation and evaluated by
SPSS 13.0 (SPSS, Inc., Chicago, IL, USA). Differences between
groups were analyzed by one way analysis of variance. Repetitive
measure analysis of variance was used in analyzing the quality of
life at varying time-points. P<0.05 was considered to indicate a
statistically significant difference.
Results
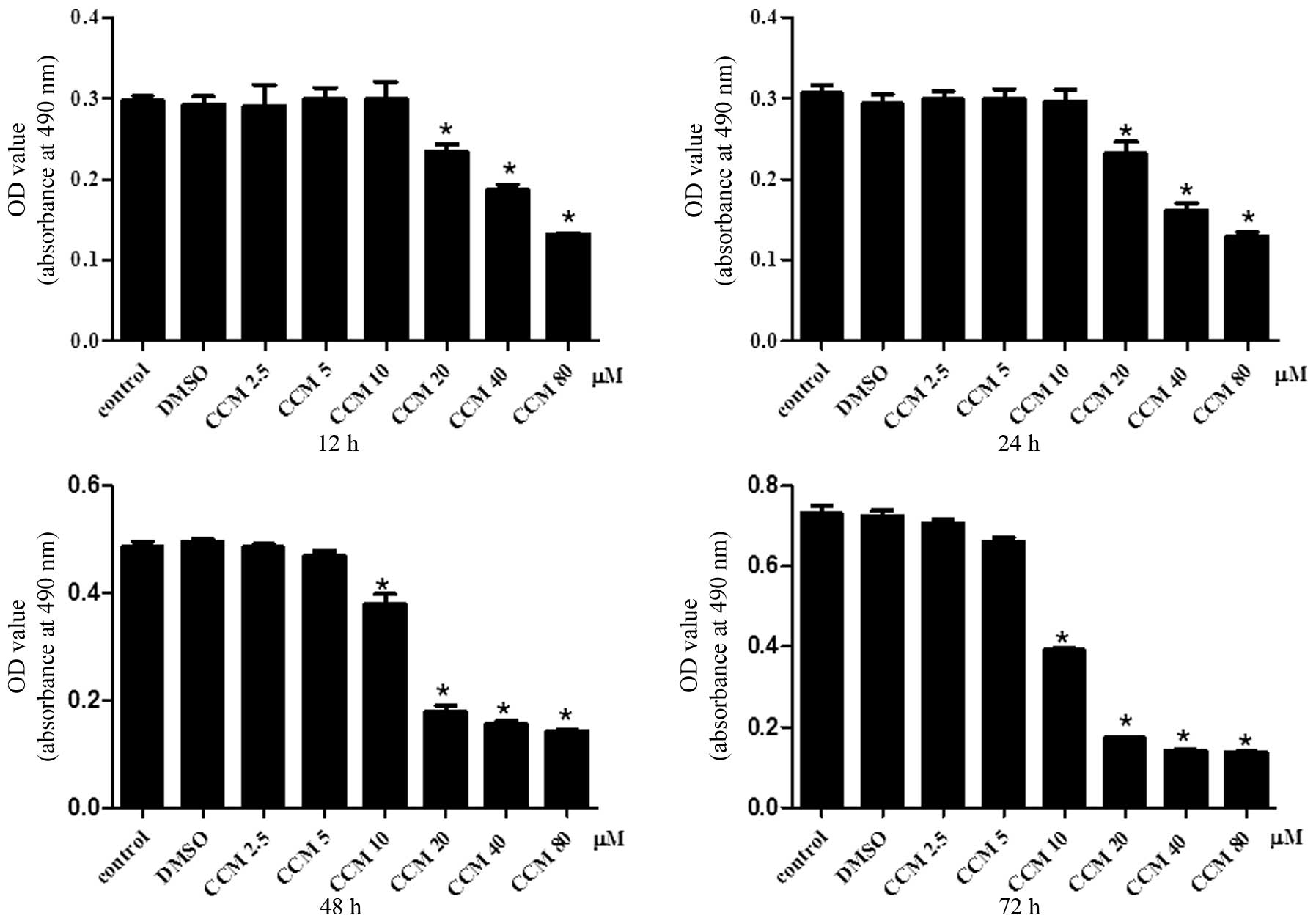
CCM inhibits the proliferation of U251
cells
The effect of CCM on U251 cell proliferation was
determined using the MTS assay. The U251 cells were treated with
CCM at increasing doses (2.5, 5, 10, 20, 40 and 80 μM) and time
intervals (12, 24, 48 and 72 h). CCM inhibited the proliferation of
the U251 cells in a dose- and time-dependent manner, as shown in
Fig. 1.
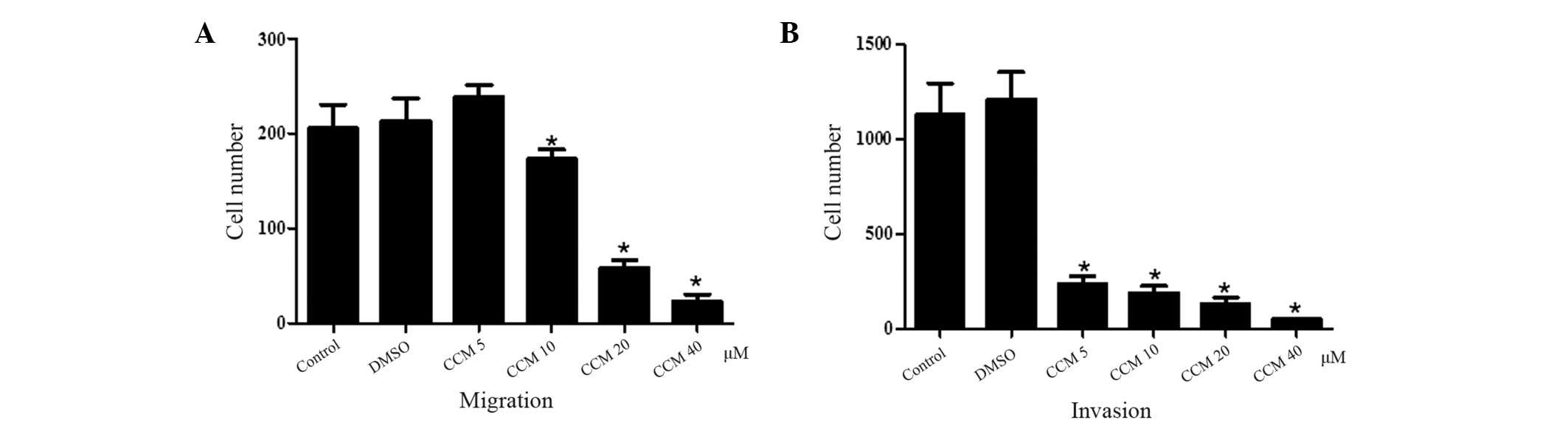
CCM inhibits the migration and invasion
of U251 cells
The effect of CCM on U251 cell migration and
invasion was determined using the Transwell Boyden chamber assay.
The migration (Fig. 2A) and
invasion (Fig. 2B) of the U251
cells were significantly decreased in a concentration-dependent
manner following CCM treatment.
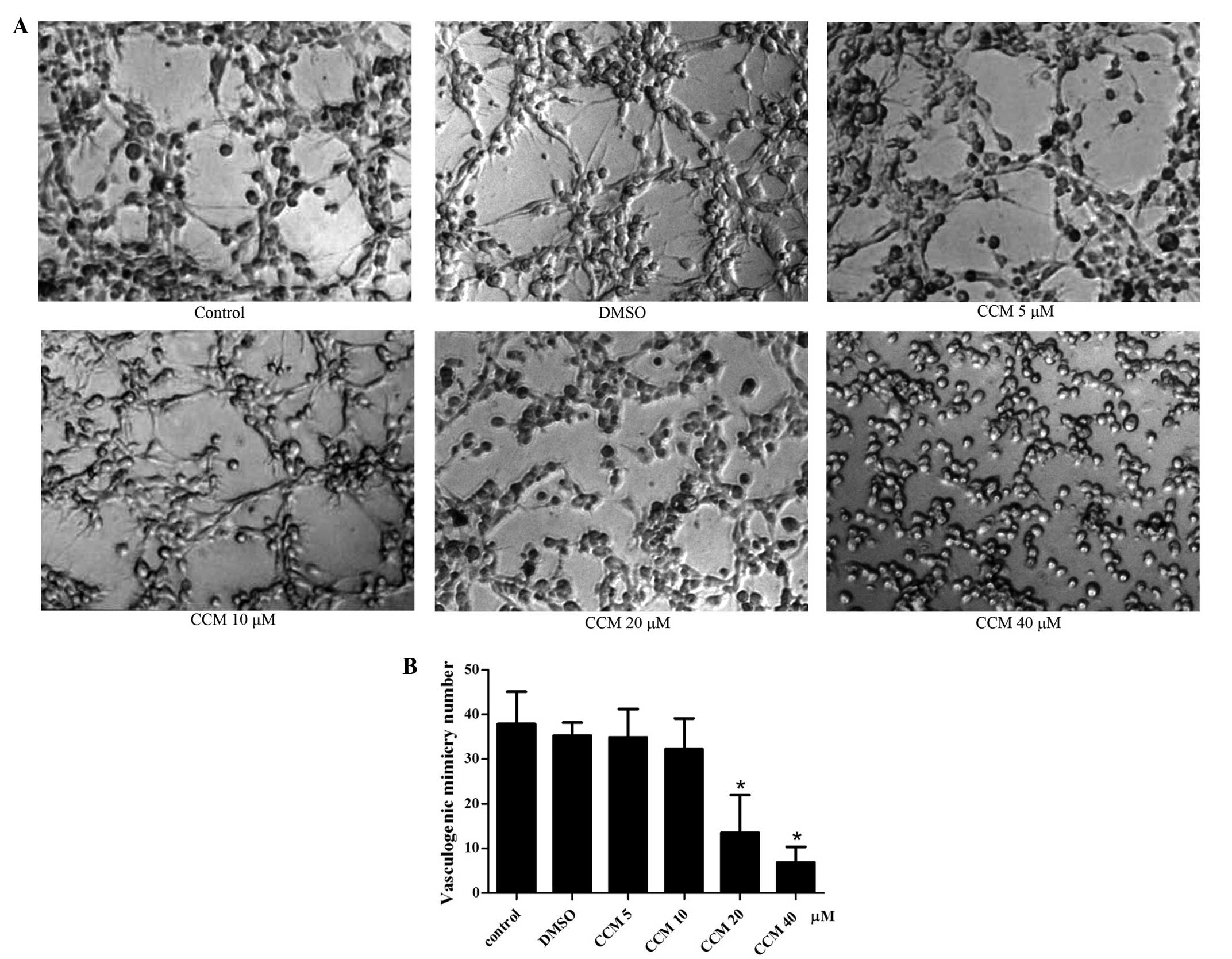
CCM inhibits VM formation of U251
cells
In complete medium (control group), the U251 cells
gradually grew and adhered, finally connecting with each other and
forming tubular network structures on Matrigel, which were judged
as the formation of VM. The most typical VM structure were formed
at 24 h, The mean number of VM structures per field was 37.88±7.28.
Therefore, the 24 h time-point was selected for the observation of
the CCM treatment. Next, the U251 cells were treated with curcumin
of increasing dose (5, 10, 20 and 40 μM) for 24 h, and the tubular
network structures (VM) were observed and quantified. CCM inhibited
the VM formation of the U251 cells in a dose-dependent manner, as
shown in Fig. 3. When incubated
with CCM, the U251 cells lost their ability to form the network
structure, the network structures on the Matrigel were disrupted
and the number of structures decreased. As the dose of CCM
increased, the number of structures gradually decreased. At the
doses of 5 and 10 μM, CCM inhibited the VM formation slightly, but
with no significant difference compared with the control group. At
the dose of 20 μM, CCM markedly inhibited the formation of the VM
structure by the U251 cells; this was significantly different
compared with control group (P<0.05). At 40 μM CCM, the network
structures on the Matrigel were disrupt almost completely
(P<0.05).
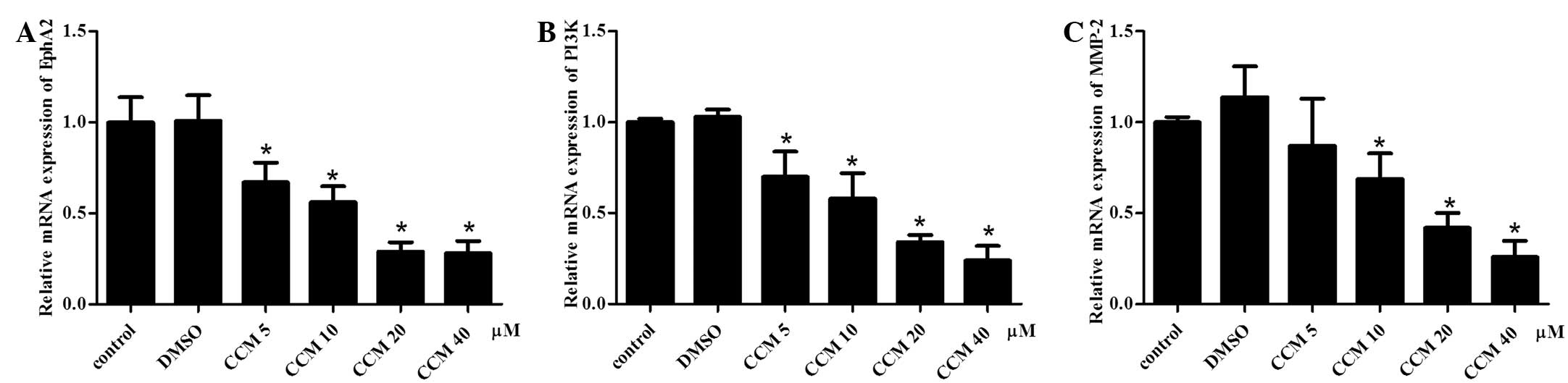
CCM suppresses the mRNA expression of
EphA2, PI3K and MMP-2 in U251 cells
The mRNA expression of EphA2, PI3K and MMP-2
following CCM treatment were detected with quantitative PCR. GAPDH
served as an internal reference. The mRNA expression of EphA2, PI3K
and MMP-2 in the CCM groups (5, 10, 20 and 40 μM) was
downregulated. The CCM suppressed the mRNA expression of EphA2,
PI3K and MMP-2 in the U251 cells in a dose-dependent manner, as
shown in Fig. 4. As the dose of CCM
increased, the mRNA expression of EphA2, PI3K and MMP-2 decreased.
At the doses of 10, 20 and 40 μM, CCM markedly suppressed the mRNA
expression of EphA2, PI3K and MMP-2, respectively; these levels
were significantly different compared with the control group
(P<0.05).
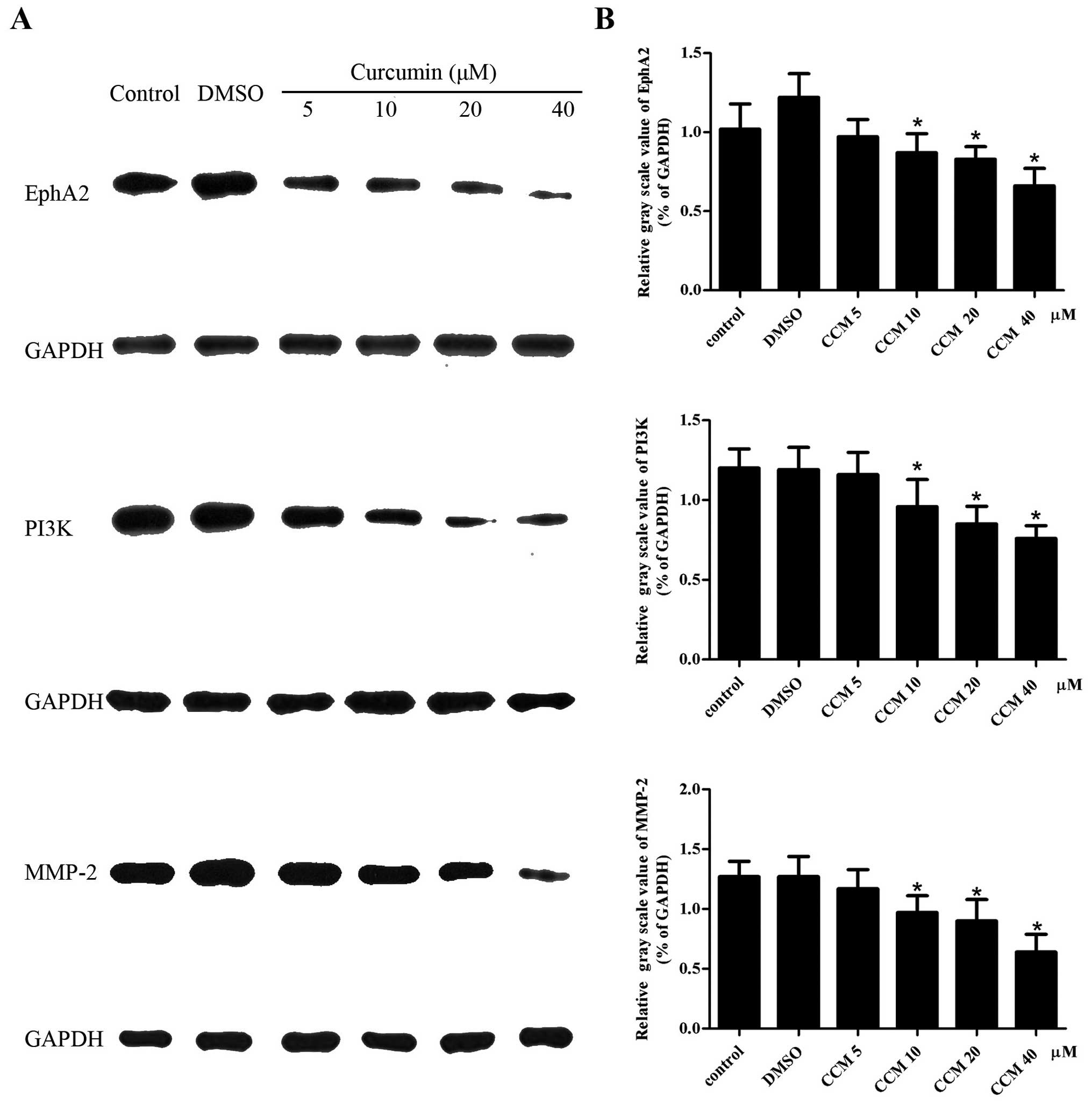
CCM decreases the protein level of EphA2,
PI3K and MMP-2 in U251 cells
The protein levels of EphA2, PI3K and MMP-2 in the
U251 cells were detected by western blot assays. GAPDH served as an
internal reference. The protein levels of EphA2, PI3K and MMP-2 in
the CCM groups (5, 10, 20 and 40 μM) were decreased. CCM suppressed
the expression of the EphA2, PI3K and MMP-2 proteins in a
dose-dependent manner, as shown in Fig.
5. At the doses of 10, 20 and 40 μM, CCM markedly decreased the
protein level of EphA2, PI3K and MMP-2, respectively; these levels
were significantly different compared with control group
(P<0.05).
Discussion
CCM has become a focus of attention due to its
inhibitory role in GBM. CCM has been found to suppress the
initiation, proliferation, migration and metastasis of glioma cells
in vitro and in vivo. The effective mechanisms of CCM
include cell cycle arrest, the induction of apoptosis, the
suppression of oncogenes and the enhancement of tumor suppressor
genes (18–22). Molecular targets, including the
PI3K/Akt/mTOR, protein kinase C, Ras/mitogen-activated protein
kinase, Wnt and intrinsic or extrinsic apoptosis pathways have been
studied (23). In the present
study, CCM suppressed the proliferation of the U251 cells in a
dose- and time-dependent manner, as determined by MTS assay. The
Transwell Boyden chamber assays showed that the migration and
invasion of the U251 cells were significantly impaired in a
concentration-dependent manner following CCM treatment. Moreover,
downregulation of the EphA2, PI3K and MMP-2 expression was
associated with the inhibition of GBM by CCM.
Inhibition of angiogenesis is one of the important
mechanisms of action for CCM against GBM. At present, studies on
the effect of CCM against GBM angiogenesis mainly focus on
endothelium-dependent angiogenesis (24). The mechanism of CCM against GBM
angiogenesis involves a number of signaling pathways and effective
molecules. CCM has been shown to inhibit the expression of VEGF
(25). CCM also inhibits the
constitutive activation of the PI3K/Akt pathway, which is
upregulated in glioma cell lines (22). Abnormal MMP expression is
significant in malignant glioma invasion into the surrounding
normal brain tissues. It has previously been shown that CCM has
broad-spectrum inhibitory activity against MMP gene expression in
human astroglioma cells. CCM has also been demonstrated to inhibit
the expression of MMP-2 and -9 in human astroglioma cells (26,27).
Since there is cross-talk between the aforementioned
pathway-related molecules and the VM formation-related molecules,
the question of whether CCM can inhibit the formation of VM in GBM
has arisen. If CCM can inhibit endothelium-dependent angiogenesis
and the formation of VM in GBM, it can become an ideal
anti-angiogenesis drug for treating GBM.
In the current study, it was reported that CCM
inhibited the VM formation of the U251 cells in a dose-dependent
manner. When incubated with CCM, the U251 cells lost their ability
to form the network structure, the network structures on the
Matrigel were disrupted and the number of structures decreased. As
the dose of CCM increased, the inhibition of VM formation also
increased. At the doses of 10, 20 and 40 μM, CCM markedly inhibited
the formation of the VM structure by the U251 cells; these levels
were significantly different compared with the control group
(P<0.05).
Subsequently, the present study explored the
mechanism of CCM inhibition on the VM formation of the U251 cells.
We hypothesized that EphA2, PI3K and MMP-2 may be the critical
factors in VM formation in GBM. Therefore, EphA2, PI3K and MMP-2
were examined by quantitative PCR and western blot assays following
CCM treatment in the U251 cells. It was found that the mRNA
expression levels of EphA2, PI3K and MMP-2 in the CCM groups (5,
10, 20 and 40 μM) were downregulated, and that the protein
expression levels were decreased. In the U251 cells, CCM suppressed
the expression of EphA2, PI3K and MMP-2 mRNA and protein in a
dose-dependent manner.
A growing body of evidence has suggested that EphA2,
PI3K and MMP-2 play significant roles in the formation of VM in
GBM. Firstly, EphA2, PI3K and MMP-2 are highly expressed in GBM,
and their expression levels are positively correlated with the
pathological grade, proliferation and invasion of GBM.
Immunohistochemical studies have revealed that the presence of VM
is associated with the expression of MMP-2, MMP-14, EphA2 and
laminin 5γ2 in medulloblastoma (4).
The PI3K/AKT signaling pathway regulates glioma cell proliferation
and invasion, while pharmacological inhibitors of PI3K/Akt
signaling (LY294002 and A443654) reduce the motility of GBM cells
and diminish the level of MMP-2 activity and MMP-2 expression
(28,29). Penta-acetyl geniposide has been
shown to exhibit an inhibitory effect on the abilities of adhesion
and motility in C6 glioma cells, decreased the expression of MMP-2
and inhibited the expression of PI3K protein (30).
Secondly, EphA2, PI3K and MMP-2 may take part in the
formation of VM in GBM. Wu et al (31) found that ectopic expression of
miR-26b inhibited the proliferation, migration and invasion of
human glioma cells. In addition, miR-26b inhibited the VM processes
accompanied with the downregulation of EphA2 proteins, which
revealed that EphA2 was correlated with the capability of VM
formation in gliomas. Ling et al (17) blocked the expression of MT1-MMP, a
cell surface activator of MMP-2-pro in glioma, by small interfering
(si)RNA transient transfection in U251MG, and found that the VM
formation was significantly impaired and that the activity of MMP-2
in the MT1-MMP siRNA group was significantly decreased. However,
Ling et al (17) also showed
that EphA2 may not be involved in TGFβ-induced VM formation. In the
current study, it was found that the expression levels of EphA2,
PI3K and MMP-2 were inhibited by CCM treatment, which was
accompanied with a decrease in VM formation in the U251 cells. This
result was similar to the results of studies on melanoma, and
indicated that the expression of EphA2, PI3K and MMP-2 may be
responsible for the capability of VM formation in gliomas (16). Future studies should confirm these
observations in vivo.
In conclusion, the present study demonstrated for
the first time that CCM inhibits the VM formation of U251 cells and
downregulates the expression of EphA2, PI3K and MMP-2. The results
provide new evidence for the use of CCM in the treatment of
malignant glioma, and may contribute to the angiogenesis-targeted
therapy for this disease.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81272806), the Natural Science
Foundation of Guangdong Province (S2012010009088) and the Projects
of the Guangzhou Municipal Health Bureau (20122A011016). The
authors would like to thank Dr Yi Arial Zeng (Chinese Academy of
Sciences, Shanghai) for providing kind advice with regard to the
editing of the original manuscript.
References
|
1
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: a clinical review. JAMA. 310:1842–1850.
2013.
|
|
2
|
Iwamoto FM and Fine HA: Bevacizumab for
malignant gliomas. Arch Neurol. 67:285–288. 2010.
|
|
3
|
Reardon DA, Desjardins A, Peters K, et al:
Phase II study of metronomic chemotherapy with bevacizumab for
recurrent glioblastoma after progression on bevacizumab therapy. J
Neurooncol. 103:371–379. 2011.
|
|
4
|
Liu XM, Zhang QP, Mu YG, et al: Clinical
significance of vasculogenic mimicry in human gliomas. J
Neurooncol. 105:173–179. 2011.
|
|
5
|
Wang SY, Yu L, Ling GQ, et al:
Vasculogenic mimicry and its clinical significance in
medulloblastoma. Cancer Biol Ther. 13:341–348. 2012.
|
|
6
|
Chen Y, Jing Z, Luo C, Zhuang M, Xia J,
Chen Z and Wang Y: Vasculogenic mimicry-potential target for
glioblastoma therapy: an in vitro and in vivo study. Med Oncol.
29:324–331. 2012.
|
|
7
|
Hess AR, Seftor EA, Gardner LM, et al:
Molecular regulation of tumor cell vasculogenic mimicry by tyrosine
phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer
Res. 61:3250–3255. 2001.
|
|
8
|
Seftor RE, Seftor EA, Koshikawa N, et al:
Cooperative interactions of laminin 5 gamma2 chain, matrix
metalloproteinase-2, and membrane type-1-matrix/metalloproteinase
are required for mimicry of embryonic vasculogenesis by aggressive
melanoma. Cancer Res. 61:6322–6327. 2001.
|
|
9
|
Hess AR, Seftor EA, Seftor RE and Hendrix
MJ: Phosphoinositide 3-kinase regulates membrane Type 1-matrix
metalloproteinase (MMP) and MMP-2 activity during melanoma cell
vasculogenic mimicry. Cancer Res. 63:4757–4762. 2003.
|
|
10
|
Paulis YW, Soetekouw PM, Verheul HM,
Tjan-Heijnen VC and Griffioen AW: Signalling pathways in
vasculogenic mimicry. Biochim Biophys Acta. 1806:18–28. 2010.
|
|
11
|
Kirschmann DA, Seftor EA, Hardy KM, et al:
Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic
and therapeutic implications. Clin Cancer Res. 18:2726–2732.
2012.
|
|
12
|
Li L, Aggarwal BB, Shishodia S, Abbruzzese
J and Kurzrock R: Nuclear factor-kappaB and IkappaB kinase are
constitutively active in human pancreatic cells, and their
down-regulation by curcumin (diferuloylmethane) is associated with
the suppression of proliferation and the induction of apoptosis.
Cancer. 101:2351–2362. 2004.
|
|
13
|
Sharma RA, Gescher AJ and Steward WP:
Curcumin: the story so far. Eur J Cancer. 41:1955–1968. 2005.
|
|
14
|
Singh S and Khar A: Biological effects of
curcumin and its role in cancer chemoprevention and therapy.
Anticancer Agents Med Chem. 6:259–270. 2006.
|
|
15
|
Das T, Sa G, Saha B and Das K: Multifocal
signal modulation therapy of cancer: ancient weapon, modern
targets. Mol Cell Biochem. 336:85–95. 2010.
|
|
16
|
Chen LX, He YJ, Zhao SZ, et al: Inhibition
of tumor growth and vasculogenic mimicry by curcumin through
down-regulation of the EphA2/PI3K/MMP pathway in a murine choroidal
melanoma model. Cancer Biol Ther. 11:229–235. 2011.
|
|
17
|
Ling G, Wang S, Song Z, et al:
Transforming growth factor-β is required for vasculogenic mimicry
formation in glioma cell line U251MG. Cancer Biol Ther. 12:978–988.
2011.
|
|
18
|
Senft C, Polacin M, Priester M, Seifert V,
Kögel D and Weissenberger J: The nontoxic natural compound Curcumin
exerts anti-proliferative, anti-migratory, and anti-invasive
properties against malignant gliomas. BMC Cancer. 10:4912010.
|
|
19
|
Weissenberger J, Priester M, Bernreuther
C, Rakel S, Glatzel M, Seifert V and Kögel D: Dietary curcumin
attenuates glioma growth in a syngeneic mouse model by inhibition
of the JAK1,2/STAT3 signaling pathway. Clin Cancer Res.
16:5781–5795. 2010.
|
|
20
|
Su CC, Wang MJ and Chiu TL: The
anti-cancer efficacy of curcumin scrutinized through core signaling
pathways in glioblastoma. Int J Mol Med. 26:217–224. 2010.
|
|
21
|
Aoki H, Takada Y, Kondo S, Sawaya R,
Aggarwal BB and Kondo Y: Evidence that curcumin suppresses the
growth of malignant gliomas in vitro and in vivo through induction
of autophagy: role of Akt and extracellular signal-regulated kinase
signaling pathways. Mol Pharmacol. 72:29–39. 2007.
|
|
22
|
Zanotto-Filho A, Braganhol E, Edelweiss
MI, et al: The curry spice curcumin selectively inhibits cancer
cells growth in vitro and in preclinical model of glioblastoma. J
Nutr Biochem. 23:591–601. 2012.
|
|
23
|
Rekers NH, Sminia P and Peters GJ: Towards
tailored therapy of glioblastoma multiforme. J Chemother.
23:187–199. 2011.
|
|
24
|
Chatterjee S and Bhattacharjee B: Use of
natural molecules as anti-angiogenic inhibitors for vascular
endothelial growth factor receptor. Bioinformation. 8:1249–1254.
2012.
|
|
25
|
Gururaj AE, Belakavadi M, Venkatesh DA,
Marmé D and Salimath BP: Molecular mechanisms of anti-angiogenic
effect of curcumin. Biochem Biophys Res Commun. 297:934–942.
2002.
|
|
26
|
Kim SY, Jung SH and Kim HS: Curcumin is a
potent broad spectrum inhibitor of matrix metalloproteinase gene
expression in human astroglioma cells. Biochem Biophys Res Commun.
337:510–516. 2005.
|
|
27
|
Perry MC, Demeule M, Régina A, Moumdjian R
and Béliveau R: Curcumin inhibits tumor growth and angiogenesis in
glioblastoma xenografts. Mol Nutr Food Res. 54:1192–1201. 2010.
|
|
28
|
Han L, Yang Y, Yue X, et al: Inactivation
of PI3K/AKT signaling inhibits glioma cell growth through
modulation of β-catenin-mediated transcription. Brain Res.
1366:9–17. 2010.
|
|
29
|
Kwiatkowska A, Kijewska M, Lipko M, Hibner
U and Kaminska B: Downregulation of Akt and FAK phosphorylation
reduces invasion of glioblastoma cells by impairment of MT1-MMP
shuttling to lamellipodia and downregulates MMPs expression.
Biochim Biophys Acta. 1813:655–667. 2011.
|
|
30
|
Huang HP, Shih YW, Wu CH, et al:
Inhibitory effect of penta-acetyl geniposide on C6 glioma cells
metastasis by inhibiting matrix metalloproteinase-2 expression
involved in both the PI3K and ERK signaling pathways. Chem Biol
Interact. 181:8–14. 2009.
|
|
31
|
Wu N, Zhao X, Liu M, et al: Role of
microRNA-26b in glioma development and its mediated regulation on
EphA2. PLoS One. 6:e162642011.
|