Introduction
Pendred syndrome, also termed goiter-deafness
syndrome, is a relatively rare recessive genetic disease, which is
characterized by congenital deafness and progressive clinical
enlargement of a goiter. Pendred syndrome was first recognized by
Vaughan Pendred, a British physician, in 1896 (1). A single mutant recessive gene,
SLC26A4(PDS), which encodes the protein pendrin, is considered to
be responsible for the goiter and deafness. Previous studies have
indicated that this protein functions as a chloride/iodine pump
(2). A lack of awareness of this
disease in clinical practice often results in cases of misdiagnosis
or missed diagnoses. Thus, the details of a typical patient with
Pendred syndrome are presented. Clinicians must raise awareness
regarding the disease and ensure correct diagnosis and
treatment.
The present report discusses a patient with a goiter
and a hearing impairment, expounding the characteristics of Pendred
syndrome so as to raise the awareness of the disease and ensure the
correct diagnosis and treatment. The patient provided written
informed consent.
Case report
A 26-year-old male presented to the Tianjin Medical
University General Hospital (Tianjin, China) due to a painless lump
in the neck, which had been inadvertently identified 6 years
previously. The lump had recently begun to exhibit rapid growth and
the patient reported symptoms of agitation, irritability,
hyperhidrosis and discomfort whilst swallowing. Prior to
presentation at Tianjin Medical University General Hospital
(Heping, China), the patient had been diagnosed with nodular goiter
by physicians at other hospitals. The patient had intermittently
taken Euthyrox® for numerous years. The patient reported
a hearing impairment and learning difficulties with language in
childhood, however, a clinical examination showed that his mental
and physical development was normal.
Physical examination showed a visible goiter, grade
III according to the World Health Organisation and Pan American
Health Organization criteria (3).
The thyroid was enlarged, and felt tough with no palpable nodules
and the thyroid boundary was clear. Laboratory tests revealed a
free thyroxine level of 9.99 pmol/l (normal rannge, 11.5–23.5
pmol/l), a free triiodothyronine level of 6.07 pmol/l (normal
range, 3.5–6.5 pmol/l) and a thyroid stimulating hormone level of
2.401 μlU/ml (0.3–5.0 μlU/ml). The thyrotrophin receptor,
thyroglobulin and thyroperoxidase antibodies, carcinoembryonic
antigen, thyroglobulin and calcitonin levels were within the normal
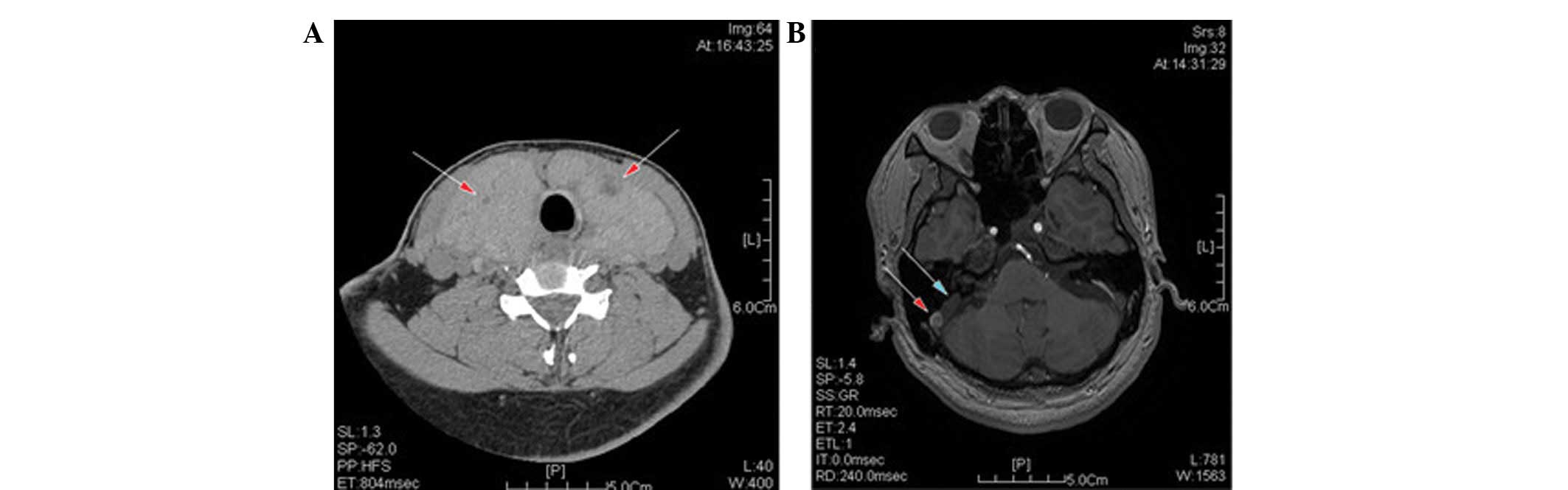
ranges. A chest X-ray revealed a narrowing of the trachea at the
level of the superior aperture of the thorax, and a thyroid
ultrasound showed multiple hypoechoic signals in each lobe and
isthmus of the thyroid. Furthermore, a cervical lymph node
enlargement was revealed (Fig. 1).
The manifestations of the cervical computed tomogrophy scan
included a diffuse enlargement of each lobe, and a thickening of
the isthmus of the thyroid gland. Numerous, oval, low- and slightly
high-density nodules were identified in the thyroid. Electrical
capacitance tomography results were consistent with the signs of a
nodular goiter. Magnetic resonance imaging of the inner ear showed
that the two vestibular aqueducts were enlarged, more markedly in
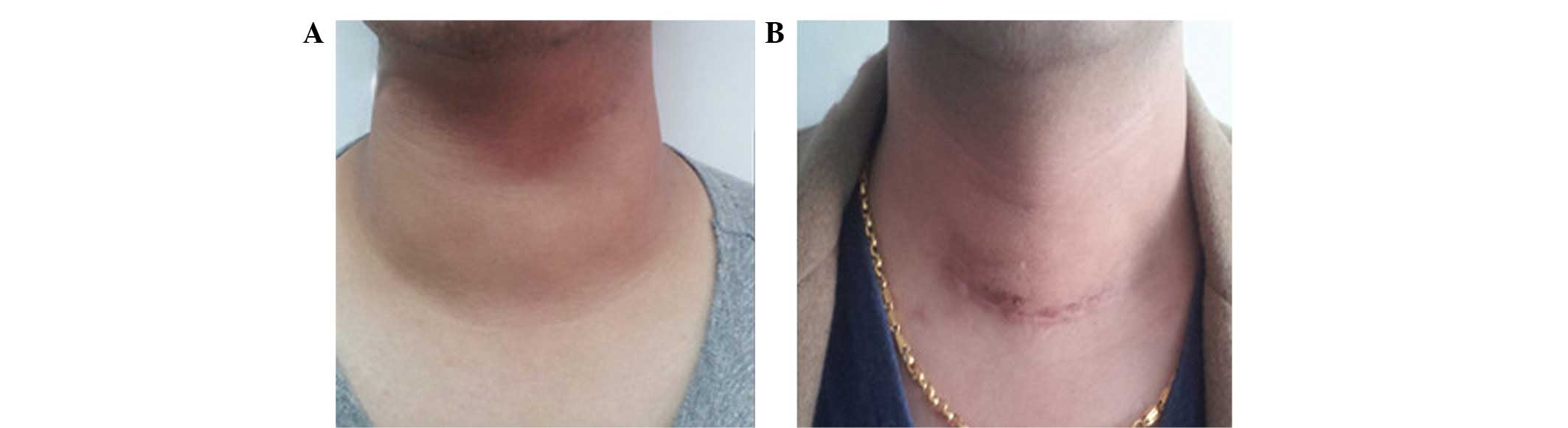
the right aqueduct. The surgical procedure was performed under
general anesthesia with an endotracheal intubation. A left
lobectomy and a subtotal right lobectomy were performed and the
patient experienced an uneventful postoperative period. Post
surgery, the patient was administered permanent hormone replacement
therapy with thyroxine (Fig. 2).
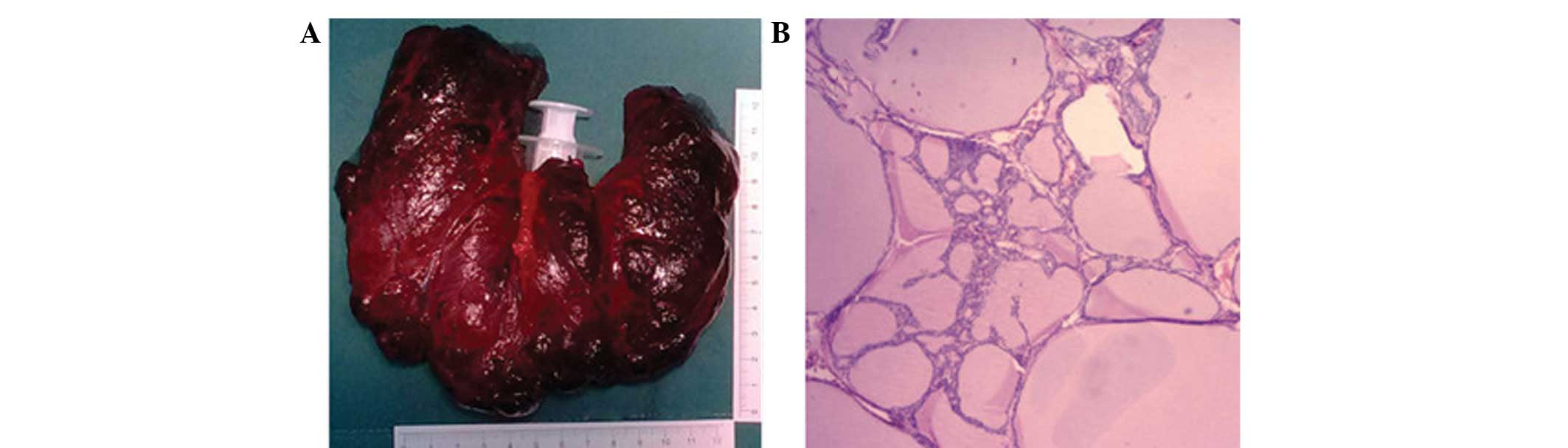
The thyroid pathology following the thyroidectomy is shown in
Fig. 3.
Discussion
Pendred syndrome is an autosomal recessive disorder,
which is characterized by familial goiter, congenital deafness and
organic iodine deficiency disorders (4). The most prominent clinical symptom in
patients with Pendred syndrome is bilateral congenital
sensorineural deafness (5). The
degree of deafness may differ among individual patients, and may
manifest at birth or appear gradually (6). The majority of patients have a speech
disorder, and a small number may experience tinnitus and vertigo
(7). The relevant imaging
examinations in patients with Pendred syndrome show that the
majority of patients exhibit a vestibular aqueduct enlargement,
with or without inner ear malformation. Furthermore, patients
commonly present with a dilated lymph sac and lymphatic vessels
(8). A typical inner ear
deformation, termed Mondini malformation, is an abnormality of the
cochlear that is characterized by an abnormally short and flat
cochlear structure (9). Goiter is
another feature of Pendred syndrome that typically develops in
patients following the onset of deafness. The early stages of
goiter is diffuse, which subsequently and gradually develops into
multiple nodular goiter. The common clinical sign of goiter in
young patients is diffuse enlargement. In adults, the thyroids are
typically enlarged with multiple palpable nodules, but with no
tremors or vascular murmurs. It has previously been reported that
Pendred syndome has the potential to become cancerous (10). Furthermore, goiter is exhibited to
varying degrees in patients with Pendred syndrome, however, it is
most significant between the ages of 20 and 30 years (8). The molecular mechanisms of Pendred
syndrome have been determined by previous studies. The pathogenic
gene, SLC26A4 (also termed PDS) is located on chromosome
7q31 (11). The PDS gene
encodes the pendrin protein, a highly hydrophobic transmembrane
protein that consists of 780 amino acids (12). A previous study demonstrated that
this protein is highly expressed in the thyroid, inner ear and
kidney (13). Pendrin functions as
a chloride/iodide pump in the thyroid, and is responsible for
transporting iodine out of the cell and into the follicular colloid
(2). A PDS gene mutation may
cause abnormal protein expression and affect iodine transport. As a
result, patients may develop a clinically enlarged thyroid. Pendrin
is expressed in the inner ear for the transport of
chloride-formate, which has a critical role in the maintenance of a
stable endolymph environment. PDS mutations may, therefore,
result in an abnormal pendrin structure and influence its chloride
ion transport function in the inner ear. Furthermore, abnormal
pendrin structure and function leads to an increase in the internal
pressure of the vestibular aqueduct and lymphatic vessels, which
increases the pressure of the internal ear. As a result, patients
are clinically characterized with a hearing impairment and even
deafness (14).
Pendred syndrome was first described by Pendred
(1) in 1896. Brian et al
reported the case of an inbred family from London, comprising of 12
siblings who were identified to suffer from the goiter-deafness
syndrome (15). It was concluded
from the observations that the disease may be associated with a
recessive gene mutation (16).
Despite previous studies, the majority of clinicians are unfamiliar
with the disease, resulting in frequent misdiagnoses. Furthermore,
patients with Pendred syndrome present with different clinical
manifestations, including varying degrees of hearing impairment and
thyroid dysfunction, which complicates diagnosis (Dhariry). The
perchlorate discharge test has traditionally been used to diagnose
Pendred syndrome, however, it is not a specific diagnostic test
(17). Patients are administered 10
mg/kg perchlorate, which is taken orally, and Iodine-131
(131I) uptake rates are measured 1 h prior to and
following administration. A positive test is indicated by an
131I uptake rate of >10%. The perchlorate discharge
test facilitates the diagnosis of Pendred syndrome, however, it is
not considered to be the gold standard. The majority of patients
exhibit euthyroid goiter, however, certain patients exhibit signs
of hypothyroidism (6). In the
present case report, a patient with Pendred syndrome was presented,
who had no family history of deafness or goiter. The patient had
experienced progressive hearing damage since early childhood and,
from adolescence, had gradually developed goiter and shown signs of
hypothyroidism. Genetic testing is a specific diagnostic method for
detecting PDS mutations, however, it is currently difficult
to perform in clinical practice. As a result, clinical
manifestations and imaging methods are more commonly adopted to
determine the diagnosis. There is currently no effective treatment
for Pendred syndrome. Adequate thyroid hormone replacement therapy
should be prescribed early to prevent further development of
goiter. In patients without oppressive symptoms and probable
canceration, surgery is not recommended. However, in the present
case the patient underwent a left lobectomy and a subtotal right
lobectomy due to the large size of the goiter and tracheal
compression, which was caused by rapid growth.
In order to manage the goiter of Pendred syndrome,
patients require adequate assessment, including observation of
clinical symptoms, as well as imaging examinations of the thyroid.
Due to the persistence of pathogenic factors, surgical removal of
sections of the gland may lead to goiter recurrence
postoperatively. As aforemtioned, surgery must be avoided in
patients without oppressive symptoms and suspicious canceration. In
patients with the aforementioned symptoms surgical intervention is
of great significance as due to the persistence of pathogenic
factors the surgical removal of the gland may lead to postoperative
goiter recurrence. Early intervention measures can be taken to
prevent disease progression and reduce thyroid growth, thereby
reducing the likelihood of obstruction and canceration.
In conclusion, it is important to determine an early
diagnosis of Pendred syndrome; however, reducing the rate of missed
diagnoses and misdiagnoses requires further investigation, thus, it
is essential for clinicians to improve their understanding of
Pendred syndrome. Misdiagnosis may be reduced, in part, by
screening for suspected cases, obtaining a full report of the
family history and performing relevant imaging examinations. The
incidence of the disease may be reduced via genetic analysis of the
disease-causing PDS gene, genetic counseling and eugenic
prenatal detection. The clinical understanding of Pendred syndrome,
as well as the molecular and genetic research based on clinical
diagnosis, will have a vital role in the prevention, early
detection, diagnosis and treatment of this disease.
References
|
1
|
Pendred V: Deaf mutism and goiter. Lancet.
148:5321896.
|
|
2
|
Kandasamy N, Fugazzola L, Evans M, et al:
Life-threatening metabolic alkalosis in Pendred syndrome. Eur J
Endocrinol. 165:167–170. 2011.
|
|
3
|
Hazarika NC and Mahanta J: Environmental
iodine deficiency and goiter prevalence in a block area of the
North Eastern region: a retrospective analysis. J Hum Ecol.
15:113–117. 2004.
|
|
4
|
Kopp P, Pesce L and Solis-S JC: Pendred
syndrome and iodide transport in the thyroid. Trends Endocrinol
Metab. 19:260–268. 2008.
|
|
5
|
Reardon W, Coffey R, Phelps PD, et al:
Pendred syndrome --100 years of underascertainment. QJM.
90:443–447. 1997.
|
|
6
|
Fagazzloa L, Cerutti N, Mannavola D, et
al: Differential diagnosis between Pendred and pseudo-Pendred
syndromes: clinical, radiologic, and molecular studies. Pediatr
Res. 51:479–484. 2002.
|
|
7
|
Kandasamy N, Fugazzola L, Evans M, et al:
Life-threatening metabolic alkalosis in Pendred syndrome. Eur J
Endocrinol. 165:167–170. 2011.
|
|
8
|
Sugiura M, Sato E, Nakashima T, et al:
Long-term follow-up in patients with Pendred syndrome: vestibular,
auditory and other phenotypes. Eur Arch Otorhinolaryngol.
262:737–743. 2005.
|
|
9
|
Griffith AJ, Telian SA, Downs C, et al:
Familial Mondini dysplasia. Laryngoscope. 108:1368–1373. 1998.
|
|
10
|
Nosé V: Thyroid cancer of follicular cell
origin in inherited tumor syndromes. Adv Anat Pathol. 17:428–436.
2010.
|
|
11
|
Massa G, Jaenen N, de Varebeke SI, et al:
Solitary thyroidnoduleas presenting syndrome of Pendred syndrome
caused by a novel splice-site mutation in intron 8 of the SLC26A4
gene. Eur J Pediate. 162:674–677. 2003.
|
|
12
|
Bizhanova A and Kopp P: Genetics and
phenomics of Pendred syndrome. Mol Cell Endocrinol. 322:83–90.
2010.
|
|
13
|
Dhahiry JS, Niamey TK, Daher AK and Biati
RK: Pendred’s syndrome; a case report and review of literatures.
The Medical Journal of Basrah University. 29:40–42. 2011.
|
|
14
|
De La Vieja A, Gnter CS and Carrasco N:
Molecular analysis of a congenital iodide transport defect: G543E
impairs maturation and trafficking of the Na+/I- symporter. Mol
Endocrinol. 19:2847–2858. 2005.
|
|
15
|
Derarmaeker R: Congenital deafness and
goiter. Am J Hum Genet. 8:253–256. 1956.
|
|
16
|
Scott DA, Wang R, Kreman TM, et al: The
Pendred syndrome gene encodes a chloride-iodide transport protein.
Nat Genet. 21:440–443. 1999.
|
|
17
|
Reardon W, Coffey R, Pembrey ME, et al:
Pitfalls in practice- diagnosis and misdiagnosis in Pendred
syndrome. J Audiol Med. 6:1–9. 1997.
|