Introduction
Hemophagocytic lymphohistiocytosis (HLH), also
termed hemophagocytic syndrome, is an aggressive hyperinflammatory
condition characterized by prolonged fever, cytopenias and
hepatosplenomegaly, as well as hemophagocytosis by activated,
morphologically benign macrophages. There are two main types of
HLH, familial HLH (FHLH) and secondary HLH. Familial HLH (FHLH) is
an autosomal recessive syndrome with an estimated prevalence of
1/50,000 live births (1). Secondary
HLH is a well-recognized entity and is associated with infections,
autoimmune diseases, immune deficiencies, metabolic diseases, drugs
or malignancies (2). The incidence
of secondary HLH is unknown. Malignancy-associated hemophagocytic
lymphohistiocytosis is mostly accompanied by lymphoid neoplasms.
The present study describes a rare case of this syndrome in
combination with acute myeloblastic leukemia (AML), in a patient
with an abnormal karyotype, who was successfully treated with
chemotherapy. Written informed consent was obtained from the the
family of the patient.
Case report
Case presentation
A 61-year-old female presented to the Hematological
Department of Navy General Hospital (Beijing, China) with a history
of high fever for 30 days. The complete blood count showed
pancytopenia; the white blood cell (WBC) count was
1.92×109/l (normal range, 4–10×109/l), the
hemoglobin (Hgb) levels were 60 g/l (normal range, 120–150 g/l)and
the platelet (PLT) count was 33×109/l (normal range,
100–300×109/l). Elevated levels of serum ferritin
(11,966.7 μg/l; normal range, 11–360 μg/l) were detected. A reduced
level of fibrinogen (Fg; 0.87 g/l; normal range, 1.5–4.0 g/l) and
increased levels of lactate dehydrogenase (LDH; 1,617 U/l; normal
range, 230–460 U/l), triglycerides (2.28 mmol/l; normal range,
1.7–2.25 mmol/l) and D-Dimer (6,779 μg/l; normal range, 0–300 μg/l)
were detected. Serum antibody to Epstein-Barr virus (EBV) were
negative, while serum antibodies to human immunodeficiency virus
(HIV), hepatitis A, B and C (HAV, HBV and HCV), tubercle bacillus
and hemococcidium were negative. Repeated blood cultures were
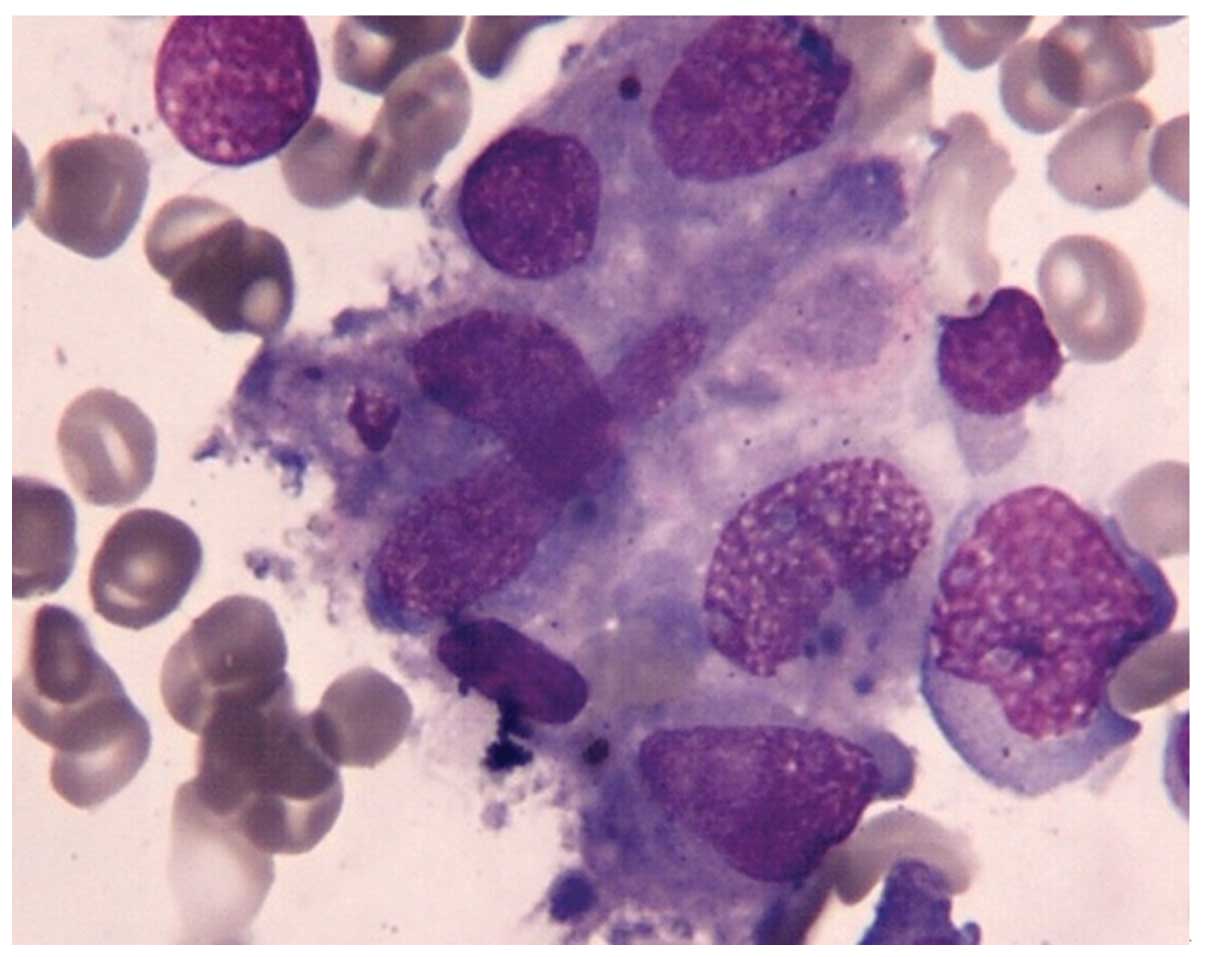
negative. Bone marrow (BM) aspirate showed increased histiocytes
(4.5%) with hemophagocytosis, and dysplasia in granulocytic and
erythroid lineage. BM examination revealed a hypercellular marrow
with 29% blasts accompanied by histiocytes with hemophagocytosis,
but the erythroid progenitors were only 0.5% of total nucleated
cells (Fig. 1). Morphology and flow
cytometry studies showed no evidence of hematological malignancy.
The levels of serum soluble interleukin 2 (IL-2) receptor (sCD25)
in plasma of the BM were 44,000 pg/ml (normal levels, <6,400
pg/ml), while natural killer (NK) cell activity was 6.72% (normal
range, 31.54–41.58%). G-banding analysis showed that these blasts
had a chromosomal abnormality with 48,X,
add(X)(p11),+3,der(7)t(1;7)(q11;p22),inv(12)(q15q24),add(14)(q32),t(14;19)(q32;q13),+18,add(21)(q22.
CT scan examination revealed splenomegaly.
Diagnosis
On the basis of both clinical and laboratory
findings (fever, splenomegaly, cytopenias, hypofibrinogenemia,
hemophagocytosis in the BM, hyperferritinemia, raised serum sCD25
levels and decreased NK-cell activity), a diagnosis of
hemophagocytic lymphohistiocytosis HLH was therefore established. A
bone marrow biopsy revealed a hypocellular marrow, which was
composed of 25% myeloblasts, and thus the patient was
simultaneously diagnosed with AML-M2.
Treatment
The patient received chemotherapy for AML,
comprising daunorubicin (40 mg/m2 i.v., days 1–3) and
cytosine arabinoside (100 mg/m2, 1-h intravenous
infusion, days 1–7). However, the treatment for HLH, according to
the HLH 2004 protocol (6), was not
started. Following the initial cycle of chemotherapy, clinical
symptoms subsided. BM examination showed resolution of
hemophagocytosis, although significant dyserythropoiesis was noted
in the BM smears. The WBC and PLT counts, as well as the LDH and Fg
levels, normalized. Hgb levels were also raised, but did not
recover to normal levels. The serum ferritin levels declined
gradually, but remained elevated (758 μg/l). The hemophagocytic
syndrome was ameliorated after the first cycle of chemotherapy.
The patient achieved BM remission without
hemophagocytosis after the second cycle of chemotherapy. Following
consolidation therapy, comprised of mitoxantrone (4
mg/m2 i.v., days 1–3) and cytosine arabinoside (100
mg/m2, 1-h infusion, days 1–7), the patient developed
pneumonia with fever. The patient succumbed to septic shock 4
months following the initial diagnosis of AML.
Discussion
HLH has been traditionally classified as either
primary familial HLH, with a genetic etiology, or secondary HLH,
which is associated with malignancies, autoimmune diseases and
infections (1). HLH is
characterized by uncontrolled cytokine production secondary to
underlying defective NK cell activity, resulting in persistent
cytotoxic T-cell activation, macrophage proliferation and
hemophagocytosis (1). Studies of
cytokine levels in the blood and tissues of HLH patients have
indicated persistently elevated levels of multiple pro-inflammatory
cytokines during symptomatic disease, including IL-1β, tumor
necrosis factor-α, IL-6, IL-8 and interferon gamma (1,4,5). It is
currently considered that hypercytokinemia and hyperchemokinemia
underlie the potentially fatal organ dysfunction in patients with
secondary HLH. Elevated levels of sCD25, a marker of T-cell
activity, have been shown to be correlated with the prognosis of
HLH in children (6). In accordance
with the International Histiocyte Society guidelines (7), five of the following eight diagnostic
criteria are required for a diagnosis of secondary HLH: Fever,
cytopenia of two cell lines, hypertriglyceridemia and/or
hypofibrinogenemia, hyperferritinemia (>500 g/l),
hemophagocytosis, elevated sCD25 levels, decreased NK cell
activity, and splenomegaly. All criteria, with the exception of
hemophagocytosis, were present in our patient.
Although a diagnosis of HLH was made in the present
case, the cause remained unclear. Secondary HLH is frequently
associated with an infectious etiology, including EBV,
cytomegalovirus, HAV, HCV, HBV, herpes simplex virus, HIV,
Escherichia coli, histoplasma and pneumocystis (8). The wide range of triggers of HLH has
prompted researchers to stress the importance of identifying the
underlying cause to enable targeted therapy (9). However, the present patient was
extensively investigated for viral, bacterial and fungal
infections, both in peripheral blood and BM samples, and was not
found to have any of the described infections. HLH has been
described in association with various types of hematological
malignancies, particularly T-cell lymphoma, although there were
proven or suspected infectious triggers in both cases (10,11).
There have been two reported cases of AML and secondary HLH, one of
which was associated with infection, another of which was
therapy-related (12,13). However, infection- and
therapy-associated HLH were thoroughly excluded in our patient.
HLH is a poor prognostic factor for patients with
hematological cancer (14–16). It is possible that the development
of leukemia in patients with genetic HLH mutations may trigger
overt HLH, particularly when combined with infections. The patient
in this case presented with chromosomal abnormalities, which may
have induced the HLH and AML. Patients with abnormalities in FHLH
genes may have defective immune surveillance of abnormal clones
and, as a result, may be predisposed to leukemia (17), as is likely in the present case.
There is no consensus on the treatment of HLH when it is
concomitant with AML. Jordan et al recommend firstly
initiating immunochemotherapy aimed at controlling the
inflammation, and then administering disease-specific therapy once
inflammatory markers have normalized (13). However, in the patient in the
present case, the AML was treated first, which in turn resulted in
resolution of the hemophagocytosis.
In conclusion, single chemotherapy for AML with HLH
was proven to be effective in the present case. The present case
report has demonstrated successful chemotherapeutic treatment of a
patient with AML-associated HLH. However, there are few reported
cases of AML occurring with HLH in the literature and, therefore,
this finding requires further investigation in a similar
setting.
References
|
1
|
Janka GE: Hemophagocytic syndromes. Blood
Rev. 21:245–253. 2007.
|
|
2
|
Janka G, Imashuku S, Elinder G, Schneider
M and Henter JI: Infection- and malignancy-associated
hemophagocytic syndromes. Secondary hemophagocytic
lymphohistiocytosis. Hematol Oncol Clin North Am. 12:435–444.
1998.
|
|
3
|
Imashuku S and Teramura T: Hemophagocytic
syndrome associated with herpes virus infections. Nihon Rinsho.
64(Suppl 3): 663–667. 2006.(In Japanese).
|
|
4
|
Szyper-Kravitz M: The hemophagocytic
syndrome/macrophage activation syndrome: a final common pathway of
a cytokine storm. Isr Med Assoc J. 11:633–634. 2009.
|
|
5
|
Hotchkiss RS and Nicholson DW: Apoptosis
and caspases regulate death and inflammation in sepsis. Nat Rev
Immunol. 6:813–822. 2006.
|
|
6
|
Henter JI, Horne A, Aricó M, et al:
HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic
lymphohistiocytosis. Pediatr Blood Cancer. 48:124–131. 2007.
|
|
7
|
Meki A, O’Connor D, Roberts C and Murray
J: Hemophagocytic lymphohistiocytosis in chronic lymphocytic
leukemia. J Clin Oncol. 24:e685–e687. 2011.
|
|
8
|
Komp DM, McNamara J and Buckley P:
Elevated soluble interleukin-2 receptor in childhood hemophagocytic
histiocytic syndromes. Blood. 73:2128–2132. 1989.
|
|
9
|
Usmani GN, Woda BA and Newburger PE:
Advances in understanding the pathogenesis of HLH. Br J Haematol.
161:609–622. 2013.
|
|
10
|
Ando K, Miyazawa K, Kuriyama Y, Kimura Y,
Mukai K and Ohyashiki K: Hemophagocytic syndrome associated with
CD8 positive T-cell chronic lymphocytic leukemia. Leuk Lymphoma.
45:193–198. 2004.
|
|
11
|
Chaker L, Segeren CM, Bot FJ and Maartense
E: Haemophagocytic syndrome and Hodgkin’s disease variant of
Richter’s syndrome after fludarabine for CLL. Eur J Haematol.
85:91–92. 2010.
|
|
12
|
Takahashi T and Matsugama M: Refractory
hemophagocytic syndrome in a patient with acute myelocytic
leukemia. Blood. 121:28202013.
|
|
13
|
Wang LX, Fei XM, Lu YL, et al: Acute
myeloid leukemia initially presenting as hemophagocytic
lymphohistiocytosis - a case report and review of the literature.
Leuk Res. 34:e46–e49. 2010.
|
|
14
|
Ishii E, Ohga S, Imashuku S, et al:
Nationwide survey of hemophagocytic lymphohistiocytosis in Japan.
Int J Hematol. 86:58–65. 2007.
|
|
15
|
Takahashi N, Chubachi A, Kume M, et al: A
clinical analysis of 52 adult patients with hemophagocytic
syndrome: the prognostic significance of the underlying diseases.
Int J Hematol. 74:209–213. 2001.
|
|
16
|
O’Brien MM, Lee-Kim Y, George TI, McClain
KL, Twist CJ and Jeng M: Precursor B-cell acute lymphoblastic
leukemia presenting with hemophagocytic lymphohistiocytosis.
Pediatr Blood Cancer. 50:381–383. 2008.
|
|
17
|
Jordan MB, Allen CE, Weitzman S,
Filipovich AH and McClain KL: How I treat hemophagocytic
lymphohistiocytosis. Blood. 118:4041–4052. 2011.
|