Introduction
Castleman’s disease (CD) is a rare
lymphoproliferative disorder with unknown origin. It is not
frequently identified in the abdomen, however, it can occur in any
region containing lymph nodes (1,2). Two
principal histological subtypes of CD have been reported:
Hyaline-vascular and plasma cell variants; additionally, there are
two clinicoradiological entities, denoted unicentric and
multicentric CD (1). CD is
exclusively diagnosed histologically and immunohistochemically
following biopsy or resectioning of the lesion. Furthermore, the
majority of unicentric cases are cured by resection of the involved
lymph node(s). Radiation or chemotherapy may be administered,
however, these treatments are not curative (1,3). The
five-year survival rate for unicentric CD is ~100% (3).
Littoral cell angioma (LCA) is also a rare disease,
and refers to a benign, vascular tumor of the spleen (4), although malignant variants have been
described (5,6). Although no age predilection has been
identified, the median age for patients with LCA is 50 years
(range, 1–77 years) and it usually occurs in adults and appears to
be extremely rare in children. Currently, a final diagnosis is only
possible by histopathological examination. LCA may be cured by with
a splenectomy (5,6).
The current study describes the clinical,
laboratory, radiological and histological findings, and the
successful treatment of a patient with concurrent CD and LCA.
Written informed consent was obtained from the patient. To date, no
cases of CD accompanied by LCA have been previously reported.
Case report
A 28-year-old female presented to the Department of
Radiology, West China Hospital (Chengdu, China) with growth
retardation, primary amenorrhea and a 10 year history of microcytic
hypochromic anemia that was resistant to iron therapy. The patient
had long-term paleness and gradually reduced physical strength. On
physical examination, the patient was 145 cm tall and exhibited no
secondary gender characteristics. In addition, moderate
hepatomegaly, splenomegaly and a nontender mass in the hypogastrium
were observed.
Laboratory test results indicated microcytic
hypochromic anemia [hemoglobin, 14 g/l (normal range, 110–150 g/l);
hematocrit, 7% (normal range, 35–45%); and mean corpuscular volume,
70.0 fl (normal range, 80–98 fl)], and thrombocytosis
(396×109/l; normal range, 100–300×109/l),
however, leukocyte count and hemoglobin electrophoresis were
normal. A bone marrow smear revealed increased erythrocytic series
(41%; normal range, 20–25%) and decreased granulocytic series
(46.5%; normal range, 50–60%). Hormone, tumor marker, hepatic and
renal function test results were all within normal ranges. An
infantile uterus and a solid mass with central calcification were
detected by pelvic ultrasonic examination. A moderately enhanced,
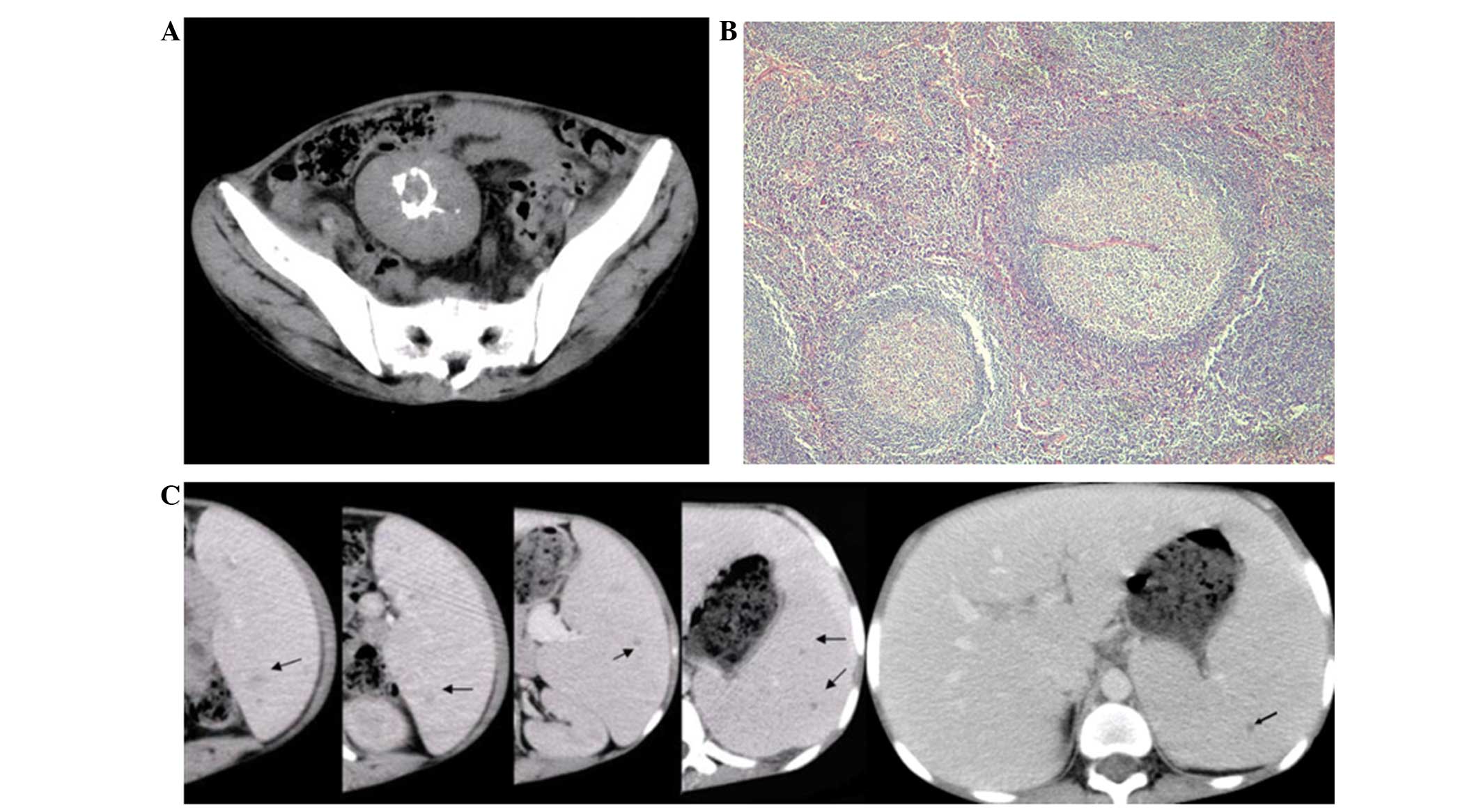
well-defined mass of 4.8×5.6 cm in size was detected at the
mesentery on computed tomography (CT) imaging; centrally located
ring-shaped calcification could also be observed. In addition,
hepatomegaly and splenomegaly were detected. Scattered small
hypo-attenuated nodules were identified in the spleen on the
arterial and portal venous phases of enhanced CT images, however,
these were isoattenuated on normal CT images (Fig. 1A and C). No abnormalities were
observed on liver parenchyma, chest or cervical CT imaging.
As the underlying nature of the mass, nodules of the
spleen and hepatomegaly could not be determined in this patient,
exploratory laparotomy was performed to remove the mass and spleen,
with additional liver biopsy. Subsequent histopathological analysis
of the mass was consistent with hyaline-vascular CD, revealing
involuting germinal centers surrounded by concentric rings of small
lymphocytes penetrated by hyalinized vessels (Fig. 1B). Immunohistochemically, the mass
was reactive for CD3±, CD20±, bcl-2 (+) and Ki-67. In addition,
pathological and immunohistochemical examination of the spleen
showed a hybrid endothelial-histiocytic phenotype, which confirmed
a diagnosis of littoral-cell angioma. Fatty degeneration was
identified in the specimen of the liver. Following surgery, the
patient’s symptoms resolved and laboratory tests normalized.
Follow-up was performed once a month, and menstruation and breast
development commenced three months following the surgery. At the
time of writing, no recurrence had been identified.
Discussion
CD was initially first described in 1954 and is also
known as angiofollicular hyperplasia or giant lymph node
hyperplasia; it is a rare disorder of lymphoid tissue (7). Little is known about the cause of this
disorder, however, it is thought to be infectious, inflammatory, or
hamartomatous in nature (1). CD may
occur in any area in which lymphoid tissue is normally found,
however, it most commonly occurs in the mediastinum (70%).
Additionally, extrathoracic sites have been reported in the neck,
axilla, mesentery, pelvis, pancreas, adrenal, and retroperitoneum
(2).
Clinically, the hyaline-vascular type of CD is
typically asymptomatic and tends to be localized, whereas the less
common plasma cell variant is occasionally associated with symptoms
including fever, anemia, weight loss, night sweats, and polyclonal
hypergammaglobulinemia, and is typically multicentric (2). The present case exhibited unique
clinical features including amenorrhea, growth retardation and
anemia. The mechanism for growth retardation in CD is unclear, and
multiple factors may be related to this, with IL-6 being one of the
important contributing factors (8).
We hypothesize that amenorrhea, which is extremely rare in CD
(9), may be associated with the
infantile uterus resulting from growth retardation. Anemia is also
extremely rare in the hyaline-vascular CD, and Yang et al
(8) proposed that elevated IL-6
acts on the IL-6 receptors in liver and tumor lymph node cells,
resulting in the enhanced expression of hepcidin, consistent with
the role of the IL-6/hepcidin pathway in anemia of chronic disease
in CD (9).
LCA, initially described in 1991, is a rare vascular
tumor, originating from the splenic littoral cells (10). Due to the limited number of cases,
the etiology of LCA remains unclear. The histopathological
characteristics of LCA include the presence of anastomosing
vascular channels lined with flat and tall endothelial cells, with
focal papillary fronds extended into the vascular channels and
normal splenic sinuses at the periphery of the lesions in the
splenic red pulp. Immunohistochemical analysis also exhibits
specific staining patterns, as the tumor shows immunoreactivity
with factor 8, CD31 and CD68, which indicates the presence of
endothelial and histocytic cells (4).
Clinically, patients with LCA may present with an
abdominal mass from splenomegaly or as an incidental finding.
Symptoms of hypersplenism (thrombocytopenia and anemia), portal
hypertension and pyrexia of unknown origin have also been
described, but are rare (5). An
association between LCA and malignancy, chronic infection or
autoimmune diseases has been proposed (6), however, the coexistence of LCA and CD
has not been previously reported in the literature. The association
between CD and LCA in the present case was unclear, however, they
have previously exhibited an association with altered immune host
response (6) thus, we hypothesized
that the coexistence of these two rare diseases in one patient may
be due to immune disorder.
Unicentric CD exhibits a number of important
features on imaging. It is usually shown to be a single, well
circumscribed, soft-tissue mass with moderate to intense
enhancement on CT, and may be homogeneous or heterogeneous
dependent on its size; additionally, low attenuation areas in the
mass has been shown to correspond to necrosis (11,12).
Intratumoral calcification, found in approximately 5–10% of CD
cases, is considered to be another characteristic CT feature of
this disease (11). CD typically
displays a variety of patterns in the abdomen and pelvis, including
punctuate, coarse, flocculent, peripheral and arborizing pattern,
which radiates from the center of the mass (2,12);
however, the central ring-shaped pattern observed in the present
case has not been previously described.
Conversely, the CT features of LCA have been well
described, with the majority being isoattenuating to slightly
hypoattenuating on non-contrast examinations. On contrast
enhancement, early hypoattenuation on arterial and most early
portal phase scans may be revealed. Heterogeneous to homogeneous
enhancement is present on the late portal phase and delayed phase.
However, a number of delayed scans have revealed a complete
contrast washout with return to isoattenuation (13). In the current study, the patient
exhibited a significantly enlarged spleen with scattered small
hypoattenuated nodules on enhanced CT, which was imperceptible on
plain CT images.
Surgical resection of the mass is usually curative
in localized CD, with dissipation of the symptoms, and splenectomy
is curative in littoral-cell angioma. However, local recurrence
following subtotal resection has also been described in localized
CD (1), as well as long-term
complications with malignancy, which has been reported in localized
CD and LCA (1,6), therefore, close clinical follow-up is
required.
In conclusion, the present study reported an unusual
case of unicentric CD accompanied by LCA, anemia, growth
retardation and amenorrhea, which was treated with surgery.
However, the relationship between unicentric CD and LCA requires
further investigation.
References
|
1
|
Jongsma TE, Verburg RJ and
Geelhoed-Duijvestijn PH: Castleman’s disease: A rare
lymphoproliferative disorder. Eur J Intern Med. 18:87–97. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim TJ, Han JK, Kim YH, Kim TK and Choi
BI: Castleman disease of the abdomen: imaging spectrum and
clinicopathologic correlations. J Comput Assist Tomogr. 25:207–214.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chronowski GM, Ha CS, Wilder RB,
Cabanillas F, Manning J and Cox JD: Treatment of unicentric and
multicentric Castleman disease and the role of radiotherapy.
Cancer. 92:670–676. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ertan G, Tekes A, Mitchell S, Keefer J and
Huisman TA: Pediatric littoral cell angioma of the spleen:
multimodality imaging including diffusion-weighted imaging. Pediatr
Radiol. 39:1105–1109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan YM, Chuah KL and Wong WK: Littoral
cell angioma of the spleen. Ann Acad Med Singapore. 33:524–526.
2004.PubMed/NCBI
|
|
6
|
Harmon RL, Cerruto CA and Scheckner A:
Littoral cell angioma: a case report and review. Curr Surg.
63:345–350. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Castleman B and Towne VW: CASE records of
the Massachusetts General Hospital Weekly Clinicopathological
Exercises: Case 40011. N Engl J Med. 250:26–30. 1954. View Article : Google Scholar
|
|
8
|
Yang M, Wang FD, Han B, et al: Castleman’s
disease presenting as prolonged anemia and growth retardation: a
case report and literature review. Acta Haematol. 125:125–129.
2011. View Article : Google Scholar
|
|
9
|
Cammisuli E, Catania V, Santuccio A and
Pennisi S: Castleman’s disease: a case report. Ann Ital Chir.
74:713–716. 2003.(In Italian).
|
|
10
|
Falk S, Stutte HJ and Frizzera G: Littoral
cell angioma: A novel splenic vascular lesion demonstrating
histiocytic differentiation. Am J Surg Pathol. 15:1023–1033. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng X, Pan K, Cheng J, Dong L, Yang K
and Wu E: Localized Castleman disease in retroperitoneum: newly
discovered features by multi-detector helical CT. Abdom Imaging.
33:489–492. 2008. View Article : Google Scholar
|
|
12
|
Meador TL and McLarney JK: CT features of
Castleman’s disease of the abdomen and pelvis. AJR Am J Roentgenol.
175:115–118. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Johnson C, Goyal M, Kim B, Wasdahl D and
Nazinitsky K: Littoral cell angioma. Clin Imaging. 31:27–31. 2007.
View Article : Google Scholar
|