Introduction
Ganglioneuroma (GN) is a benign and slow-growing
tumor that originates from the primitive neural crest cells.
Histologically, the tumor is composed of Schwann mesenchymal cells
and gangliocytes. GNs may arise in any region of the paravertebral
sympathetic plexus (1–4), but are typically observed in the
posterior mediastinum (39–43%) or the retroperitoneum (32–52%). By
contrast, the neuromas rarely occur in the adrenal glands (5,6). The
majority of patients with adrenal GNs (AGNs) do not exhibit any
clinical symptoms. Furthermore, the tumors are usually clinically
dormant and asymptomatic, even when large in size (1). Following the extensive use of imaging
procedures, such as ultrasonography and computed tomography (CT),
the number of diagnosed GNs has increased (6,7). A large
number of AGNs are identified during physical examinations or due
to the presence of other diseases. The incidence of AGN is
gradually increasing in China, due to increased awareness with
regard to early detection; and dozens of cases have been reported
in recent years (8). The majority of
previous studies concerning AGNs have been case reports, and
information regarding the pathogenesis of the disease is limited.
The establishment of a guideline for clinicians has proven
extremely challenging, and therefore, AGNs are often misdiagnosed
as a different type of tumor (8,9). The
present study describes a large AGN that was incidentally
identified in a 58-year-old female. The diagnosis was confirmed by
histopathological examination. Following diagnosis, the patient was
treated by laparoscopy. The present study also includes a review of
the relevant literature in order to provide clinicians with
information concerning this uncommon malignancy. Written informed
consent was obtained from the patient.
Case report
A 58-year-old female was admitted to the Department
of Oncology at the Affiliated Hospital of Guangdong Medical College
(Zhanjiang, Guangdong, China) in May 2011 complaining of face and
lower extremity dropsy that had been apparent for >20 days. The
patient had no symptoms of headaches, abdominal pain, nausea,
vomiting, urinary urgency or fever, and there was no loss of
appetite or weight. An ultrasound scan, which had been performed
two days previously at a local hospital, had revealed a mass in the
kidney. The patient's medical history was unremarkable. The
patient's blood pressure was within the normal range and
demonstrated no fluctuations. In addition, a physical examination
did not reveal an abnormal mass, tenderness or rebound tenderness
in the abdomen.
Laboratory tests revealed a cortisol level of 6.78
µg/dl (normal range, <10 µg/dl) and an adrenocorticotrophin
level of <10 µg/dl (normal range, <46 µg/dl). Catecholamine
metabolites in the blood and urine were within normal ranges.
Furthermore, routine tumor markers demonstrated no abnormalities
and the fecal occult blood test was negative.
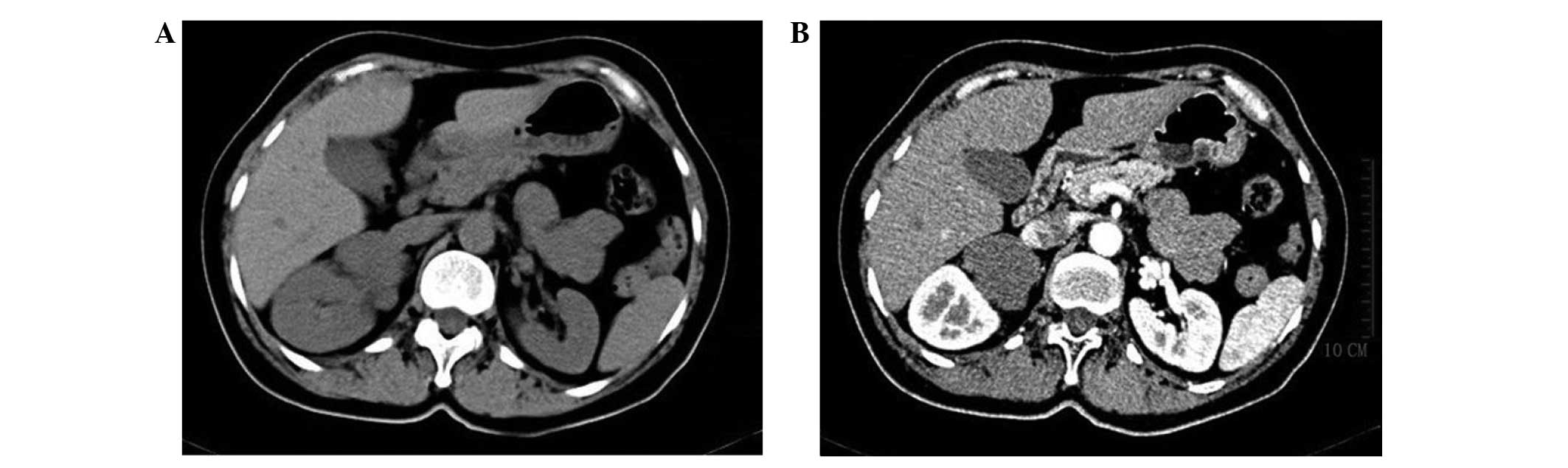
Contrast-enhanced CT of the upper abdomen revealed
an irregular and homogenous solid tumor in the right adrenal gland,
measuring ~6×4.5×7 cm3. The CT value was ~28 HU. The
arterial phase CT value was ~31 HU and the venous phase CT value
was 50 HU. The lesion and surrounding structures exhibited clear
demarcation. The left adrenal gland parenchyma was uniform in
density and demonstrated enhanced evenness, and there was no
evidence of abnormal nodes or an enhanced soft-tissue mass
(Fig. 1). Retroperitoneal lymph node
involvement was not detected, and there was no evidence of invasion
to the adjacent tissues or organs. Chest X-ray did not reveal any
abnormal lesions.
Due to the patients physical condition, a
laparoscopic complete excision of the mass was performed through a
transabdominal approach. Following surgery, pathological and
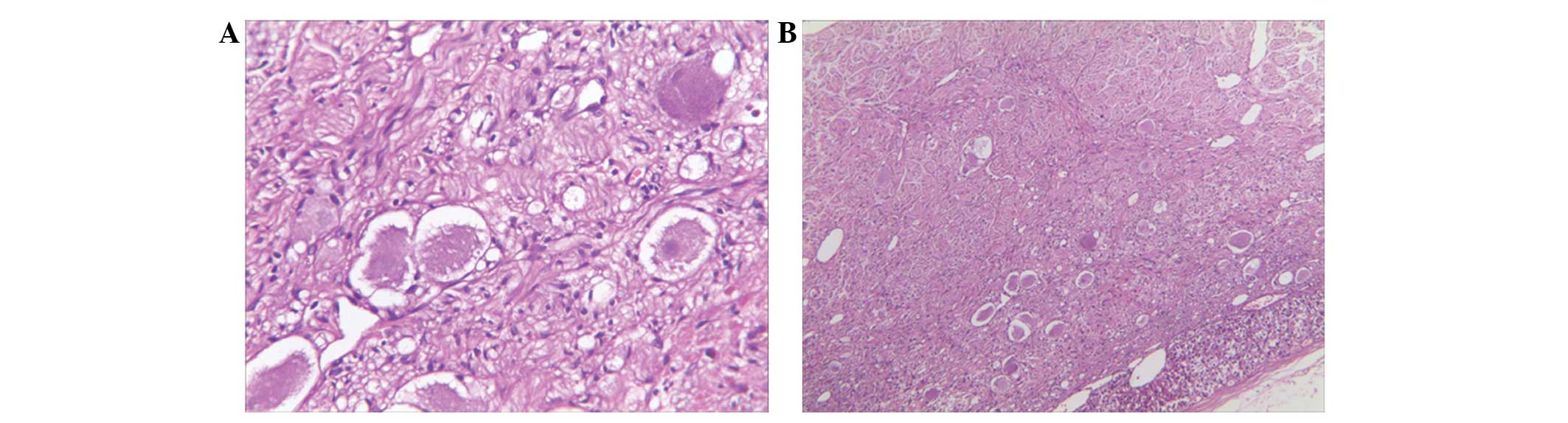
immunohistological examinations were performed. The cut surface of
the tumor was solid, whitish-gray in color, and demonstrated no
evidence of necrosis or hemorrhage. Microscopically, the sections
were composed of spindle-shaped cells that demonstrated an
irregular growth pattern and occasional scattered mature ganglionic
cells with focal lymphocytic infiltration and dystrophic
calcification (Fig. 2A and B). The
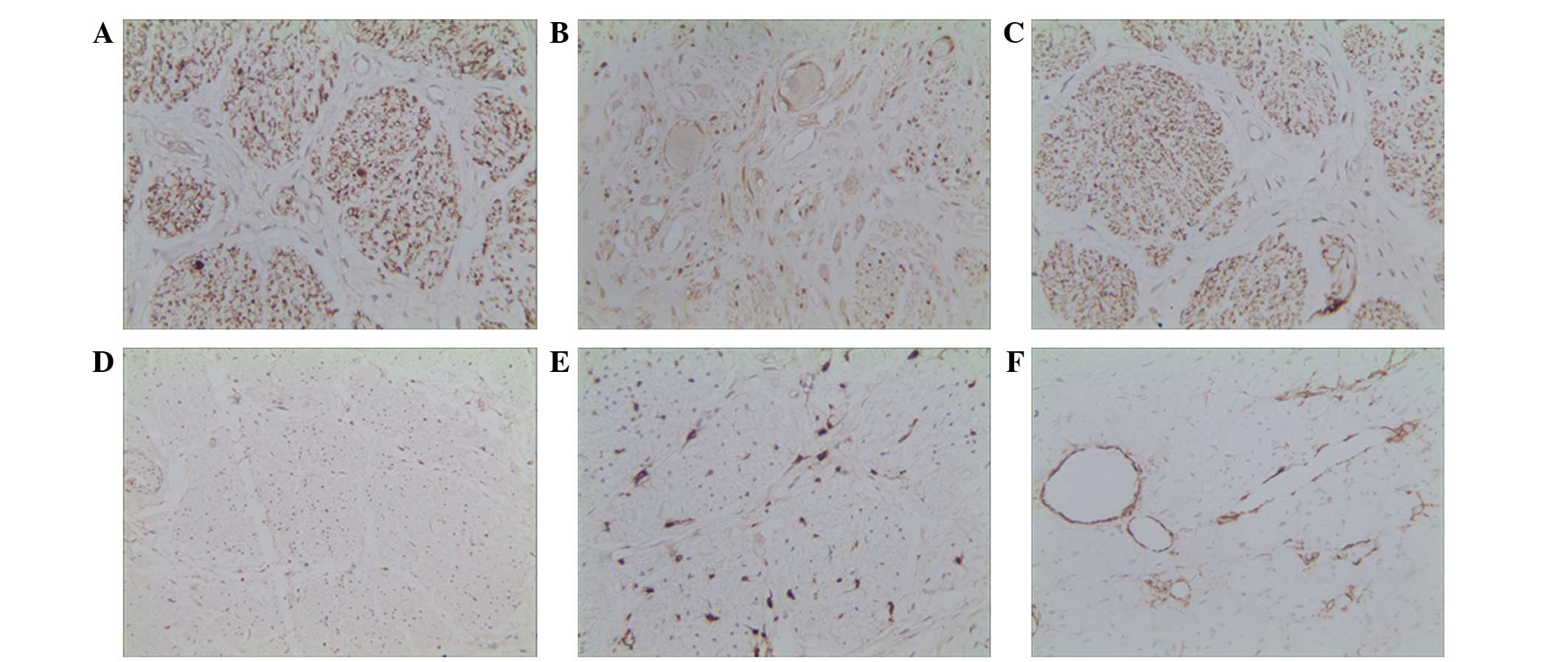
immunohistochemical analysis revealed that the ganglion cells were
positive for neuron-specific enolase (NSE; Fig. 3A), S-100 (Fig. 3B) and cluster of differentiation
(CD)56 (Fig. 3C), but negative for
cytokeratin (Fig. 3D), leukocyte
common antigen (Fig. 3E) and smooth
muscle actin (Fig. 3F). The specimen
did not exhibit any histological evidence of malignant
degeneration. These results established a histological diagnosis of
AGN.
The post-operative period was uneventful, and the
patient was discharged on the fifth day after surgery. The patient
returned to the hospital 18 months later in good health. CT did not
reveal any evidence of tumor recurrence or metastasis, and the
cosmetic result was satisfactory.
Discussion
GN originates from primitive neural crest cells,
arises in sympathetic ganglion cells and is composed of mature
Schwann cells, nerve fibers and ganglion cells (2,3,7,10). Tumors
originating from ganglion cells form a family consisting of GNs,
ganglioneuroblastomas and neuroblastomas. Ganglioneuroblastomas
exhibit intermediate differentiation, whilst neuroblastomas are
highly-malignant tumors (1–3). GNs most often occur in children and
young adults aged between 10 and 40 years old (9,11). AGN is
an extremely rare, differentiated, benign and slow-growing tumor.
In adults, it most commonly affects the right adrenal gland
(6). The majority of GN patients do
not exhibit any clinical manifestations. However, if they do, the
most common one is an asymptomatic, slow-growing abdominal mass or
abdominal pain. Due to its hormonally, non-secreting nature, an AGN
may rarely cause symptoms of diarrhea, virilization and
hypertension (2,12–14). This
phenotype is attributed to that fact that these tumors do not
secrete excess catecholamines or steroid hormones (15). The majority of patients with GNs are
hormonally silent. However, a minority of patients do produce
hormones, such as catecholamines, vasointestinal peptide and
androgens, which result in symptoms of weakness, diarrhea,
hypertension and virilization (13,14,16,17).
Upon CT, AGNs usually appear as oval or round,
homogenous and well-circumscribed masses, which often partially or
completely surround major blood vessels. AGNs develop as slightly
hypodense masses, with multiple punctate calcifications visible
internally. Enhanced CT scan of the tumor focus is without
enhancement or with only mild enhancement (2,18).
Microscopically, the tumor sections are primarily
composed of spindle cells clustered into fascicles. Mature ganglion
cells scattered or arranged in small clusters are also observed
(1). A diagnosis of GN depends upon
whether or not the tumor is composed of ganglion cells (19). Immunohistochemical results, including
S100, CD56, synaptophysin and NSE positivity, are also important
for forming a diagnosis (20).
Distinguishing between AGNs and other neurogenic
tumors, such as neuroblastomas, ganglioneuroblastomas,
pheochromocytomas and adenomas, can be challenging (1,13).
Neuroblastomas and ganglioneuroblastomas, which originate from
ganglion cells, usually occur in younger patients and children.
These tumors develop at the same sites as GNs, but are generally
more aggressive and metastasize to the bone, liver and lung in ~50%
of cases (21). Since
pheochromocytomas secrete catecholamine or steroid hormones,
patients often present with symptoms of paroxysmal hypertension
associated with palpitations, sweating and dizziness or headaches
(22). Patients also usually present
with a family history of the disease (23,24).
However, since GNs generally lack clear clinical symptoms or are
asymptomatic, patients with hypertension may require further
hormonal investigations (25).
Specific endocrine analyses should be performed as a definite
differential approach in order to detect hormone levels. An
elevated catecholamine level in a 24-h urine sample is suggestive
of a pheochromocytoma. By contrast, the same test may be negative
in patients with GNs, since these tumor cells do not secrete
catecholamine hormones (15). CT and
magnetic resonance imaging are commonly used during the formation
of a differential diagnosis. Upon CT, AGNs appear as solid, round
adrenal masses without evidence of tumor invasion. The most
prominent imaging finding of an AGN is a count of <40 HU,
whereas a significant enhancement upon CT is commonly observed in
adrenal pheochromocytomas (26). A
pathological investigation is often a conclusive diagnostic
determinant used to differentiate between these tumors. The gross
and histopathological morphological appearance and
immunohistochemical features differ significantly due to the
different origins of the tumors (20).
Laparoscopic adrenalectomy has become the gold
standard curative treatment for the majority of patients with
adrenal tumors. Due to its minimally invasive nature, laparoscopy
may a be more suitable approach to treat AGNs compared with
traditional open abdominal surgery (27,28). All
AGN patients undergo an open or laparoscopic adrenalectomy,
depending upon detailed analysis of individual criteria, taking
into consideration the tumor location, function and distance to
neighboring organs or blood vessels. Open surgery is performed in
patients with blood loss of >800 ml and a violent fluctuation in
intra-operative blood pressure (4).
Due to its benign nature, AGNs rarely metastasize to the regional
lymph nodes or distant organs (29),
and recurrence is rare following surgical resection (4). For this reason, unnecessary wide
excisions should be avoided. Distinguishing between GNs that
secrete catecholamines and other neurogenic tumors and
pheochromocytomas can be challenging. Therefore, if patients
present with a high level of catecholamines prior to surgery, a
diagnosis of pheochromocytoma should be considered.
In conclusion, forming a pre-operative diagnosis of
GN can be a formidable task, particularly in asymptomatic cases.
Therefore, early detection, diagnosis and treatment are extremely
important. It is common for AGNs to be misdiagnosed, therefore,
careful evaluations and surgical treatments are required. Diagnosis
can be extremely challenging, and can only be achieved by means of
histological evaluation. However, the prognoses of AGNs are usually
favorable, and recurrence is rare following surgical resection.
References
|
1
|
Zografos GN, Kothonidis K, Ageli C, et al:
Laparoscopic resection of large adrenal ganglioneuroma. JSLS.
11:487–492. 2007.PubMed/NCBI
|
|
2
|
Radin R, David CL, Goldfarb H and Francis
IR: Adrenal and extra-adrenal retroperitoneal ganglioneuroma:
imaging findings in 13 adults. Radiology. 202:703–707. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang CY, Hsieh YL, Hung GY, et al:
Ganglioneuroma presenting as an asymptomatic huge posterior
mediastinal and retroperitoneal tumor. J Chin Med Assoc.
66:370–374. 2003.PubMed/NCBI
|
|
4
|
Kouriefs C, Leris AC, Mokbel K, et al:
Abdominal ganglioneuromas in adults. Urol Int. 71:110–113. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ichikawa T, Koyama A, Fujimoto H, et al:
Retroperitoneal ganglioneuroma extending across the midline: MR
features. Clin Imaging. 17:19–21. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rha SE, Byun JY, Jung SE, et al:
Neurogenic tumors in the abdomen: tumor types and imaging
characteristics. Radiographics. 23:29–43. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lonergan GJ, Schwab CM, Suarez ES and
Carlson CL: Neuroblastoma, ganglioneuroblastoma, and
ganglioneuroma: radiologic-pathologic correlation. Radiographics.
22:911–934. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li L, Shao J, Gu J, et al: Adrenal
ganglioneuromas: experience from a retrospective study in a Chinese
population. Urol J. 11:1485–1490. 2014.PubMed/NCBI
|
|
9
|
Erem C, Ucuncu O, Nuhoglu I, et al:
Adrenal ganglioneuroma: report of a new case. Endocrine.
35:293–296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ambros IM, Zellner A, Roald B, et al: Role
of ploidy, chromosome 1p, and Schwann cells in the maturation of
neuroblastoma. N Engl J Med. 334:1505–1511. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Domanski HA: Fine needle aspiration of
ganglioneuroma. Diagn Cytopathol. 32:363–366. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pillinger SH, Bambach CP and Sidhu S:
Laparoscopic adrenalectomy: a 6-year experience of 59 cases. ANZ J
Surg. 72:467–470. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tischler AS: Divergent differentiation in
neuroendocrine tumors of the adrenal gland. Semin Diagn Pathol.
17:120–126. 2000.PubMed/NCBI
|
|
14
|
Juarez D, Brown RW, Ostrowski M, et al:
Pheochromocytoma associated with neuroendocrine carcinoma. A new
type of composite pheochromocytoma. Arch Pathol Lab Med.
123:1274–1279. 1999.PubMed/NCBI
|
|
15
|
Chen CL, Huang ST, Chang PL and Ng KF:
Adrenal ganglioneuroma: report of five cases. Chang Gung Med J.
23:550–554. 2000.PubMed/NCBI
|
|
16
|
Geoerger B, Hero B, Harms D, et al:
Metabolic activity and clinical features of primary
ganglioneuromas. Cancer. 91:1905–1013. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Watanabe T, Noshiro T, Kusakari T, et al:
Two cases of pheochromocytoma diagnosed histopathologically as
mixed neuroendocrine-neural tumour. Intern Med. 34:683–687. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamaguchi K, Hara I, Takeda M, et al: Two
cases of ganglioneuroma. Urology. 67:e1–e4. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Joshi VV and Silverman JF: Pathology of
neuroblastic tumors. Semin Diagn Pathol. 11:107–117.
1994.PubMed/NCBI
|
|
20
|
Rao RN, Singla N and Yadav K: Composite
pheochromocytoma-ganglioneuroma of the adrenal gland: A case report
with immunohistochemical study. Urol Ann. 5:115–118. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dubois C, Jankowski A, Gay-Jeune C, et al:
Imaging of adrenal ganglioneuroma: a case report. J Radiol.
86:659–662. 2005.[In French]. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi BB, Li HZ, Chen C, et al: Differential
diagnosis and laparoscopic treatment of adrenal pheochromocytoma
and ganglioneuroma. Chin Med J (Engl). 122:1790–1793.
2009.PubMed/NCBI
|
|
23
|
Elder EE, Edler G and Larsson C:
Pheochromocytoma and functional paraganglioma syndrome: no longer
the 10% tumor. J Surg Oncol. 89:193–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Manger WM and Eisenhofer G:
Pheochromocytoma: diagnosis and management update. Curr Hypertens
Rep. 6:477–484. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kasperlik-Zaluska AA, Roslonowska E,
Slowinska-Srzednicka J, et al: 1,111 patients with adrenal
incidentalomas observed at a single endocrinological center:
incidence of chromaffin tumors. Ann N Y Acad Sci. 1073:38–46. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maweja S, Materne R, Detrembleur N, et al:
Adrenal ganglioneuroma. A neoplasia to exclude in patients with
adrenal incidentaloma. Acta Chir Belg. 107:670–674. 2007.PubMed/NCBI
|
|
27
|
Zografos GN, Markou A, Ageli C, et al:
Laparoscopic surgery for adrenal tumors. A retrospective analysis.
Hormones (Athens). 5:52–56. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsuru N, Ushiyama T and Suzuki K:
Laparoscopic adrenalectomy for primary and secondary malignant
adrenal tumors. J Endourol. 19:702–709. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Srinivasan R, Koliyadan KS, Krishnand G
and Bhat SS: Retroperitoneal ganglioneuroma with lymph node
metastasis: a case report. Indian J Pathol Microbiol. 50:32–35.
2007.PubMed/NCBI
|