Introduction
Nitric oxide (NO) is synthesized in a variety of
tissues and displays various physiological actions via activating
soluble guanylyl cyclase (sGC), which converts guanosine
triphosphate (GTP) to cyclic guanosine monophosphate (cGMP),
thereby leading to an increase in the intracellular concentration
of cGMP (1). cGMP is an important
secondary messenger and exerts its effects through activating
cGMP-dependent protein kinases (PKGs). To date, two types of PKGs
have been identified in mammalian cells, namely PKG I and II. The
activated PKGs exert their biological effects through
phosphorylating target proteins (2,3). The
target proteins of PKGs have attracted significant attention and,
in particular, the significance of PKG II and its target proteins
is currently a major research focus. Epidermal growth factor
receptor (EGFR) is a receptor tyrosine kinase with a molecular
weight of 170 kDa. Activated EGFR may initiate the signal
transduction of several pathways, including mitogen-activated
protein kinase (MAPK) pathway-mediated proliferation (4,5). Our
previous studies demonstrated that, when PKG II is highly expressed
through infecting the cells with an adenoviral construct encoding
PKG II cDNA (Ad-PKG II), and activated by cGMP, the proliferation
of gastric cancer cells is inhibited (6,7). The
mechanism of the inhibitory effect of PKG II on the activation of
EGFR is through blocking the tyrosine phosphorylation/activation of
EGFR. During this process, the cell membrane-permeable cGMP
analogue 8-pCPT-cGMP is required to activate PKG II (8,9). NO is
synthesized by NO-synthase (NOS) or released by NO donors;
increased NO levels may activate sGC, leading to the increase of
intracellular cGMP. In the present study, the effect of the NO
donor sodium nitroprusside (SNP) and the NOS substrate L-arginine
on the activation of PKG II and the proliferative activity and
signaling of Ad-PKG II-infected gastric cancer cells was
investigated.
Materials and methods
Cell lines, antibodies and
chemicals
The AGS human gastric cancer cell line was provided
by the Institute of Cell Biology (Shanghai, China). Adenoviral
vectors encoding the cDNA of β-galactosidase (Ad-LacZ) and PKG II
(Ad-PKG II) were obtained from Dr Gerry Boss and Dr Renate Pilz
(University of California, San Diego, CA, USA). Dulbecco's modified
Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained
from Gibco-BRL (Grand Island, NY, USA). The rabbit polyclonal
antibody against PKG II was purchased from Abgent Biotechnology
(San Diego, CA, USA; catalog no. AP8001a; dilution, 1:200). The
goat polyclonal IgG antibody against phosphorylated
vasodilator-stimulated phosphoprotein (p-VASP; Ser239; catalog no.
sc-23507; dilution, 1:200) and the horseradish peroxidase
(HRP)-conjugated mouse monoclonal IgG1 antibody against
β-actin were obtained from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA; catalog no. sc-47778; dilution, 1:1,000). Mouse
anti-p-EGFR (Tyr1068) monoclonal antibody was purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA; catalog no. 2236;
dilution, 1:1,000.). Rabbit anti-p-ERK (Thr202/Tyr204) polyclonal
antibody was obtained from Bioworld Technology (St. Louis Park, MN,
USA; catalog no. BS5016; dilution, 1:500). The HRP-conjugated goat
anti-mouse, goat anti-rabbit and rabbit anti-goat polyclonal IgG
secondary antibodies (catalog nos. 115-035-003, 111-035-003 and
305-035-003, respectively; dilution, 1:10,000) were purchased from
Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The
cellular membrane-permeable cGMP analog 8-pCPT-cGMP was obtained
from Calbiochem (San Diego, CA, USA). EGF and SNP were purchased
from Sigma-Aldrich (St. Louis, MO, USA). L-arginine was obtained
from the Beyotime Institute of Biotechnology (Shanghai, China).
Cell Counting Kit-8 (CCK-8) was obtained from R&S Biotechnology
(Shanghai, China). The human cGMP ELISA kit was obtained from
Hushang Biotechnology (Shanghai, China) and the
electrochemiluminescence (ECL) reagents were purchased from
Millipore (Billerica, MA, USA).
Cell culture and treatment
The AGS cells were maintained in DMEM supplemented
with 10﹪ FBS and penicillin (100 IU/ml)/streptomycin (100 mg/ml)
and incubated at 37°C in 5﹪ CO2. The medium was changed
every 2 days and the cells were subcultured at confluence. The
cells were seeded in 6-well plates at a density of 70–80﹪ of
confluence and were infected with Ad-LacZ or Ad-PKG II (with a
multiplicity of infection of 100﹪) or mock-infected the following
day. At 24 h post-infection, the medium was replaced with
serum-free medium and the culture was continued for 12 h. The
infected cells were incubated with 250 µM 8-pCPT-cGMP for 1 h, the
NO donor or precursor (1 mM SNP for 2 h and 2 mM L-arginine for 4
h) and incubated with 100 ng/ml EGF for 5 min.
CCK-8 assay
A total of 10,000 cells (in 150 µl complete DMEM)
were seeded in one well of a 96-well plate. Following attachment,
the cells were infected with Ad-LacZ and Ad-PKG II for 24 h. The
cells were then washed and serum-starved overnight. Thereafter, the
cells were incubated in the presence of 8-pCPT-cGMP (250 µΜ) for 1
h, SNP (1 mM) for 2 h or L-arginine (2 mM) for 4 h, and then
treated with EGF (100 ng/ml) for 24 h. Approximately 10 µl of CCK-8
dye was added to each well and the plate was incubated for 30 min.
The optical density (OD) at 450 nm was measured using an ELx800
Absorbance Reader (BioTek Instruments, Inc., Winooski, VT, USA).
Data are presented as the relative proliferation, calculated by
dividing the OD value of each group by the OD value of Ad-LacZ
group.
ELISA
The differently treated cells were harvested by
phosphate-buffered saline (pH 7.2–7.4) to a concentration of 1
million/ml. The collected cells were subjected to three freeze-thaw
cycles to further break the cell membranes. Subsequently, the
samples were centrifuged for 20 min at 0.4–0.9 × g. The collected
supernatants were processed using the human cGMP ELISA kit,
according to the manufacturer's instructions. The absorbance was
determined at 450 nm using the ELx800 Absorbance Reader. The
standard curve was constructed by plotting the absorbance for each
standard on the x-axis against the concentrations on the y-axis;
the sample concentrations were calculated according to the sample
OD value and the standard curve.
Western blotting
The protein samples were subjected to SDS-PAGE
(8–12﹪) according to the molecular size of the target protein and
transferred onto a polyvinylidene difluoride (PVDF) membrane. The
PVDF membranes were blocked using 5﹪ non-fat milk in Tris-buffered
saline with Tween (TBS-T) for 1 h at room temperature. Incubation
with the primary antibodies was performed at 4°C overnight,
followed by incubation with the corresponding secondary antibodies
at room temperature for 1 h, with three washes in TBS-T (0.1% Tween
20) subsequent to each incubation. The signal was visualized using
ECL detection reagents.
Statistical analysis
Data are expressed as means ± standard deviation.
Statistical analysis was performed using a two-tailed analysis of
variance with SPSS 15.0 statistical software (SPSS, Inc., Chicago,
IL, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
SNP and L-arginine inhibit the
proliferation of AGS cells expressing high levels of PKG II
AGS cells were infected with Ad-PKG II to increase
the expression of the PKG II protein. SNP or L-arginine was added
to the culture medium to activate PKG II. The proliferative
activity of the cells was evaluated with CCK-8. The results
demonstrated that, in cells infected with Ad-LacZ (control virus)
and treated with EGF (100 ng/ml, 24 h), there was a significant
increase in proliferative activity compared with cells infected
with Ad-LacZ alone (P<0.01, Fig.
1). In cells infected with Ad-PKG II, treated with 8-pCPT-cGMP
(250 µM, 1 h) and then treated with EGF (100 ng/ml, 24 h), the
proliferative activity was markedly inhibited. In cells infected
with Ad-LacZ, treated with SNP (1 mM, 2 h) or L-arginine (2 mM, 4
h) and then with EGF (100 ng/ml, 24 h), the proliferation was
partially inhibited. However, in cells infected with Ad-PKG II,
treated with SNP (1 mM, 2 h) or L-arginine (2 mM, 4 h), and then
treated with EGF (100 ng/ml, 24 h), significant inhibition of
EGF-induced proliferation was observed, as was in cells infected
with Ad-PKG II, treated with 8-pCPT-cGMP and then with EGF
(Fig. 1). These results indicated
that, similar to the cGMP analogue 8-pCPT-cGMP, SNP and L-arginine
were able to activate PKG II and exert an anti-proliferative effect
on AGS cells highly expressing PKG II.
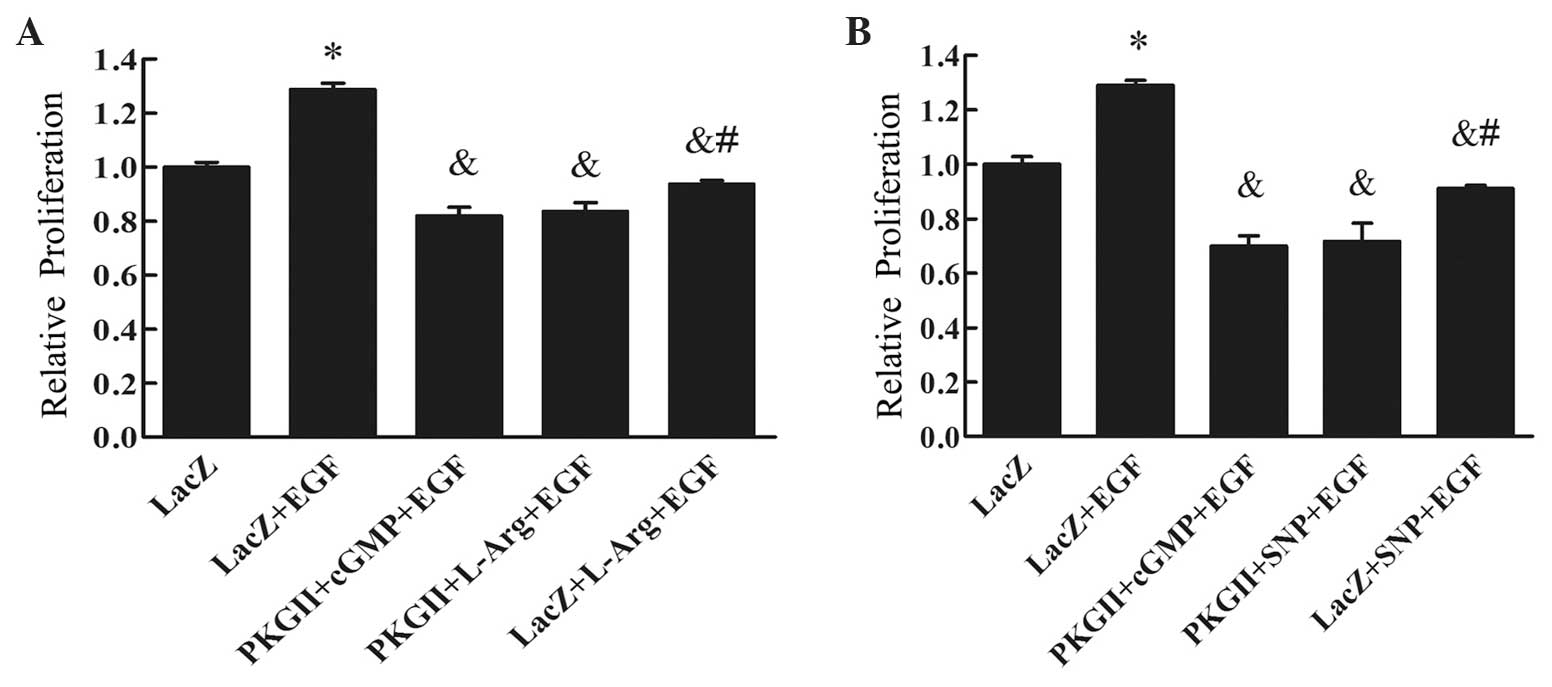 | Figure 1.Results of the CCK-8 assay for (A)
L-Arg and (B) SNP. AGS cells were infected with either Ad-LacZ or
Ad-PKG II, serum-starved for 12 h, then stimulated with 8-pCPT-cGMP
(250 µΜ) for 1 h, SNP (1 mM) for 2 h or L-Arg (2 mM) for 4 h, and
subsequently treated with EGF (100 ng/ml) for 24 h. The
proliferation of the cells was analyzed using the CCK-8 kit and the
relative proliferative activities are presented as means ± standard
deviation of three independent experiments. It was demonstrated
that EGF treatment (100 ng/ml for 24 h) promoted cell proliferation
(*P<0.01 compared with the Ad-LacZ group). The proliferative
activity of the Ad-PKG II + SNP + EGF and the Ad-PKG II + L-Arg +
EGF groups was markedly decreased (&P<0.01
compared with the Ad-LacZ + EGF group). In addition, the inhibitory
effect on proliferation of the Ad-PKG II + SNP + EGF and the Ad-PKG
II + L-Arg + EGF groups was more prominent compared with that of
the Ad-LacZ + SNP + EGF and Ad-LacZ + L-Arg + EGF groups
(#P<0.05). CCK-8, Cell Counting Kit-8; Ad-LacZ,
adenoviral vector encoding β-galactosidase cDNA; Ad-PKG II,
adenoviral vector encoding PKG II; L-Arg, L-arginine; SNP, sodium
nitroprusside; EGF, epidermal growth factor; cGMP, cyclic guanosine
monophosphate; PKG II, type II cGMP-dependent protein kinase. |
SNP and L-arginine increase the
production of cGMP in AGS cells
Once NO is released, it activates sGC, thereby
increasing the level of cGMP (10,11). In
this experiment, ELISA was performed to detect the level of cGMP in
differently treated cells. The results demonstrated that, in AGS
cells, the concentration of cGMP was increased by treatment with
SNP or L-arginine in a dose- and time-dependent manner, confirming
that SNP and L-arginine treatment increases the production of cGMP
(Table I).
 | Table I.SNP and L-arginine treatment increases
the production of cGMP in AGS cells. |
Table I.
SNP and L-arginine treatment increases
the production of cGMP in AGS cells.
| A, Effect of
treatment by dose |
|---|
|
|---|
|
| Dose (mM) |
|---|
|
|
|
|---|
| Treatment | 0 | 1 | 2 | 4 |
|---|
| SNP (2 h) | 18.15±0.3 |
20.78±0.7a |
22.94±0.6a |
24.37±0.4a |
| L-arginine (4 h) | 14.97±0.5 |
22.59±0.8a |
26.16±0.8a |
28.34±0.4a |
|
| B, Effect of
treatment by time |
|
|
| Time (h) |
|
|
|
| Treatment | 0 | 1 | 2 | 4 |
| SNP (1 mM) | 17.55±0.4 |
20.93±1.0a |
23.26±0.5a |
25.10±0.9a |
| L-arginine (2
mM) | 14.06±0.6 |
20.90±1.1a |
24.63±1.0a |
27.51±0.6a |
SNP and L-arginine increase the level
of p-VASP Ser239
High levels of cGMP may activate PKG II and cause
phosphorylation of the substrate protein of this kinase. VASP is a
substrate of protein kinases, including PKGs, and its Ser239
residue is a target specific to PKG II (12,13). In
this experiment, we analyzed SNP- and L-arginine-induced activation
of PKG II by detecting the level of p-VASP Ser239 by western
blotting. The results demonstrated that 8-pCPT-cGMP treatment (250
µM, 1 h) increased the phosphorylation of VASP at Ser239. Treatment
with SNP (1 mM, 2 h) or L-arginine (2 mM, 4 h) also caused a
significant increase of Ser239 phosphorylation of VASP (Fig. 2). These results indicated that SNP and
L-arginine treatment leads to PKG II activation.
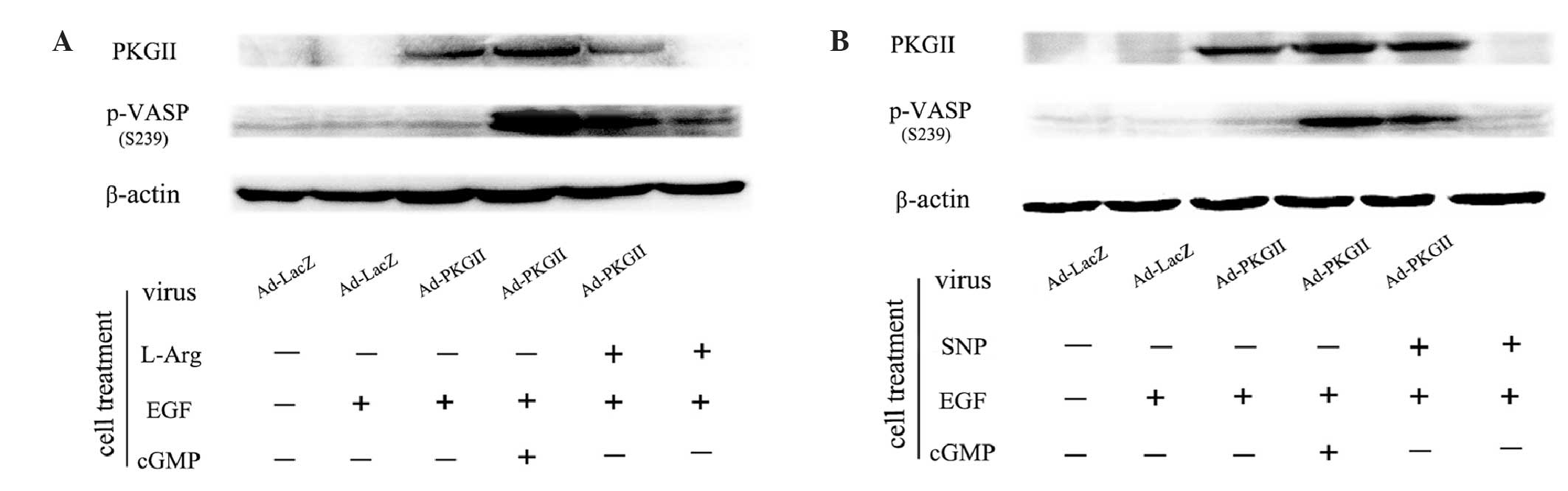 | Figure 2.(A) L-Arg and (B) SNP increased the
level of p-VASP Ser239. (A) AGS cells were infected with either
Ad-LacZ or Ad-PKG II for 24 h and then serum-starved overnight. The
Ad-LacZ group received no drug treatment. In the Ad-LacZ + EGF and
Ad-PKG II + EGF groups, the cells were treated with EGF (100 ng/ml)
for 5 min. In the Ad-PKG II + cGMP + EGF group, the cells were
incubated with 8-pCPT-cGMP (250 µΜ) for 1 h and then stimulated
with EGF (100 ng/ml) for 5 min. In the Ad-PKG II + L-Arg + EGF and
the L-Arg + EGF groups, the cells were treated with L-Arg (2 mM, 4
h) and then with EGF (100 ng/ml) for 5 min. (B) The treatments of
the former four groups were same as described in A. In the latter
two groups, the cells were incubated with 1 mM SNP for 2 h,
followed by EGF (100 ng/ml) for 5 min in the Ad-PKG II + SNP + EGF
and the SNP + EGF groups. The cells were harvested and the extracts
were subjected to western blotting with the corresponding
antibodies, as described in Materials and methods. The results are
representative of three independent experiments. L-Arg, L-arginine;
p-VASP, phosphorylated vasodilator-stimulated phosphoprotein;
Ad-LacZ, adenoviral vector encoding β-galactosidase cDNA; Ad-PKG
II, adenoviral vector encoding PKG II; SNP, sodium nitroprusside;
PKG II, type II cGMP-dependent protein kinase; EGF, epidermal
growth factor; cGMP, cyclic guanosine monophosphatase. |
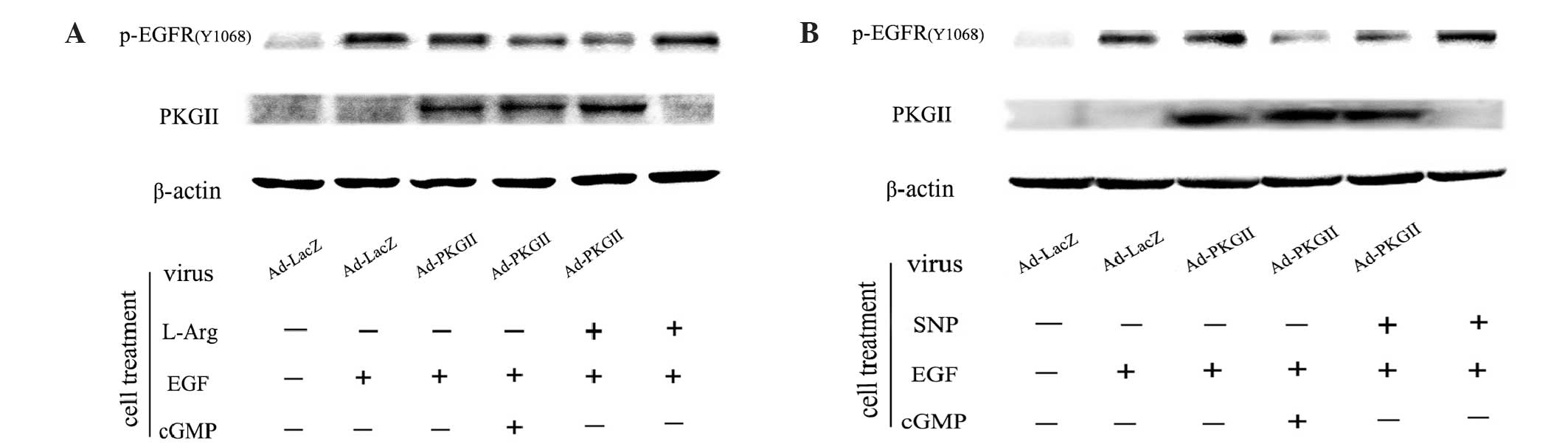
EGF-induced EGFR Tyr1068
phosphorylation is inhibited by SNP- or L-arginine-induced
activation of PKG II
As a transmembrane tyrosine kinase, EGFR is
dimerized and auto-phosphorylated by binding with ligands, such as
EGF. Tyr1068 is one of the auto-phosphorylation sites of EGFR
associated with the MAPK-mediated signaling pathway (14,15). Our
previous results demonstrated that PKG II inhibited EGF-induced
Tyr1068 phosphorylation of EGFR (7,8). The
present study investigated whether the NO donor/precursor inhibited
EGF-induced Tyr1068 phosphorylation of EGFR through activating
exogenous PKG II. The results of the western blotting revealed
that, in cells infected with Ad-LacZ and treated with EGF (100
ng/ml, 5 min), the level of Tyr1068 phosphorylation of EGFR
increased markedly (Fig. 3). In cells
infected with Ad-PKG II and treated with SNP (1 mM, 2 h) or
L-arginine (2 mM, 4 h), the EGF-induced increase of Tyr1068
phosphorylation of EGFR was efficiently inhibited. However, in
cells without Ad-PKG II infection, treatment with SNP or L-arginine
exerted no clear effect on EGF-induced Tyr1068 phosphorylation of
EGFR. Therefore, it was confirmed that NO donors and precursors
(SNP and L-arginine) inhibit EGF-induced Tyr1068 phosphorylation of
EGFR through activating PKG II.
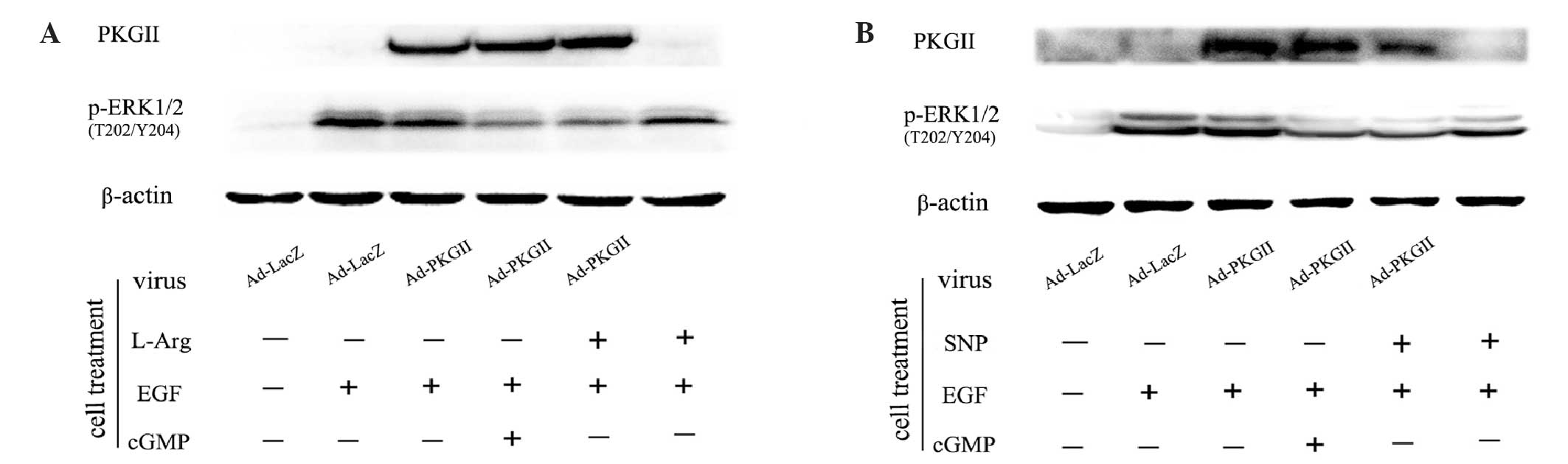
SNP and L-arginine inhibit
EGF-stimulated phosphorylation of ERK1/2 at Thr202/Tyr204
When EGFR is phosphorylated at Tyr1068, it initiates
MAPK/ERK-mediated signaling (16).
Phosphorylation of ERK1/2 at the Thr202/Tyr204 residues is one of
the key events of the signaling process of this pathway (17,18).
Western blotting with an antibody against p-ERK1/2 (Thr202/Tyr204)
was performed to detect the dual phosphorylation of ERK. The
results demonstrated that EGF treatment (100 ng/ml, 5 min) caused
an obvious increase in the level of p-ERK1/2 (Thr202/Tyr204).
However, the phosphorylation was efficiently inhibited by
pre-infecting cells with Ad-PKG II and activating the enzyme by
treatment with SNP (1 mM, 2 h) or L-arginine (2 mM, 4 h).
Furthermore, in cells without Ad-PKG II infection, SNP (1 mM, 2 h)
or L-arginine (2 mM, 4 h) treatment also exerted a mild inhibitory
effect on EGF-induced p-ERK1/2 (Fig.
4). Therefore, treatment with SNP or L-arginine alone has a
mild inhibitory effect on p-ERK1/2. However, when the expression of
PKG II was increased by Ad-PKG II infection, SNP and L-arginine
treatment completely inhibited EGF-induced phosphorylation of
p-ERK1/2 through the activation of PKG II.
Discussion
NO may be synthesized by three isoforms of NOS,
namely neuronal, inducible (in macrophages) and endothelial NOS.
Once released, NO diffuses through the cell membrane and enters the
cytosol, where it meets the target protein, such as sGC (19,20). NO
may bind to the protoporphyrin ring of sGC, causing activation of
the enzyme and leading to the formation of cGMP (21,22). As an
intracellular signaling molecule, cGMP plays an important role in
the regulation of various cellular events and may exert its effects
through PKGs, which catalyze the phosphorylation at
serine/threonine residues on substrate proteins, thereby altering
their activities (23).
In our previous experiments, it was demonstrated
that PKG II blocked the EGF-induced activation of EGFR and
inhibited EGFR-initiated signal transduction and the proliferation
of gastric cancer cells (6,7). To obtain a high activity of PKG II in
cancer cells, which usually express this enzyme at a low level,
Ad-PKG II was used to increase the expression of PKG II in these
cells. The membrane-permeable cGMP analogue 8-pCPT-cGMP was
required to activate PKG II. The present study was designed to
identify a reagent to replace 8-pCPT-cGMP.
Since NO induces cGMP production, a NO donor and a
NO precursor were selected to increase the content of NO in AGS
cells infected with Ad-PKG II, with the intention of using
endogenous cGMP to activate PKG II. The results of the ELISA
revealed that the NO donor SNP and the NO precursor L-arginine
caused a dose- and time-dependent increase of intracellular cGMP in
the cells. The effect of SNP and L-arginine on PKG II activity was
confirmed by detecting the phosphorylation of VASP, the
serine/threonine residues of which are substrates for the PKA and
PKG (24). There are three
phosphorylation sites of VASP, namely Ser157, Ser239 and Tyr278
(13,25–27). Tao
et al (13) reported that
Ser239 was a key phosphorylation site of VASP for PKG II
activation. Therefore, the level of p-VASP Ser239 was detected to
reflect PKG II activity. The results demonstrated that, under
treatment with cGMP, the level of p-VASP Ser239 was markedly
increased in cells pre-infected with Ad-PKG II. Similar to cGMP,
SNP and L-arginine also increased the level of p-VASP Ser239,
causing PKG II activation in these cells.
In the present study, the inhibitory effects of SNP
and L-arginine on EGF-induced proliferative signaling; the
proliferation of gastric cancer cells infected with Ad-PKG II was
also confirmed. When combined with EGF, EGFR is activated, which
then recruits the effectors to its phosphorylated intracellular
domain and initiates the downstream protein-mediated signaling.
Among the phosphorylation sites, Tyr1068 is associated with the
MAPK/ERK pathway (28). It was
revealed that cGMP-induced PKG II activation blocks the EGF-induced
phosphorylation of EGFR at Tyr1068. The present study revealed that
treatment with SNP or L-arginine alone did not cause a distinct
inhibition of EGF-induced Tyr1068 phosphorylation of EGFR in cells
without Ad-PKG II infection. However, when PKG II was highly
expressed following Ad-PKG II infection, SNP or L-arginine were
able to efficiently inhibit EGF-induced Tyr1068 phosphorylation of
EGFR. The effect of SNP and L-arginine on EGF/EGFR-induced
signaling of the MAPK/ERK pathway was then further investigated.
The results demonstrated that treatment with SNP or L-arginine
alone exerted a mild inhibitory effect on the EGF-induced
Thr202/Tyr204 phosphorylation of ERK1/2, which is a key signaling
event of the MAPK/ERK pathway. This was also observed by Sang et
al (29). However, when combined
with Ad-PKG II infection, SNP and L-arginine treatment markedly
inhibited the EGF-induced activation of p-ERK1/2, suggesting that
SNP and L-arginine-induced NO/cGMP production exerts an effect on
the activation of ERK, but not EGFR. However, through the
activation of PKG II, SNP and L-arginine exerted inhibitory effects
on EGFR and ERK activation and, therefore, exerted more distinct
inhibitory effects on proliferative signaling. In conclusion, a NO
donor and a NOS substrate may replace 8-pCPT-cGMP and activate PKG
II by increasing the level of endogenous cGMP, providing an
alternative method of activating this potential cancer inhibitory
factor.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81272755, 81201959 and
81001100); the Natural Science Foundation Project of Jiangsu
Province (no. 12KJB310001); China Postdoctoral Science Foundation
(no. 2014M561599); Postdoctoral Research Funding Plan in Jiangsu
Province (no. 1401144C); and the Specialized Research Fund for
Senior Personnel Program of Jiangsu University (no. 11JDG114). The
authors would like to thank Dr Gerry Boss and Dr Renate Pilz of the
University of California (San Diego, CA, USA) for providing the
adenoviral constructs.
References
|
1
|
Bohme E and Schmidt HH: Nitric oxide and
cytosolic guanylate cyclase: components of an intercellular
signalling system. Z Kardiol 78 Suppl. 6:75–79. 1989.
|
|
2
|
Orstavik S, Natarajan V, Tasken K, Jahnsen
T and Sandberg M: Characterization of the human gene encoding the
type I alpha and type I beta cGMP-dependent protein kinase (PRKG1).
Genomics. 42:311–318. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orstavik S, Solberg R, Taskén K, et al:
Molecular cloning, cDNA structure and chromosomal localization of
the human type II cGMP-dependent protein kinase. Biochem Biophys
Res Commun. 220:759–765. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wells A: EGF receptor. Int J Biochem Cell
Biol. 31:637–643. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang H, Grenley MO, Bravo MJ, Blumhagen
RZ and Edgar BA: EGFR/Ras/MAPK signaling mediates adult midgut
epithelial homeostasis and regeneration in Drosophila. Cell Stem
Cell. 8:84–95. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen YC, Ren F, Sang JR, Tao Y and Xu WR:
Type II cGMP-dependent protein kinase inhibits proliferation of the
gastric cancer cell line BGC-823. Mol Med Rep. 3:361–366. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu Y, Chen Y, Qu R, Lan T and Sang J: Type
II cGMP-dependent protein kinase inhibits EGF-triggered signal
transduction of the MAPK/ERK-mediated pathway in gastric cancer
cells. Oncol Rep. 27:553–558. 2012.PubMed/NCBI
|
|
8
|
Lan T, Chen Y, Sang J, et al: Type II
cGMP-dependent protein kinase inhibits EGF-induced MAPK/JNK signal
transduction in breast cancer cells. Oncol Rep. 27:2039–2044.
2012.PubMed/NCBI
|
|
9
|
Jiang L, Lan T, Chen Y, et al: PKG II
inhibits EGF/EGFR-induced migration of gastric cancer cells. PLoS
One. 8:e616742013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Medeiros JV, Gadelha GG, Lima SJ, et al:
Role of the NO/cGMP/K (ATP) pathway in the protective effects of
sildenafil against ethanol-induced gastric damage in rats. Br J
Pharmacol. 153:721–727. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Russo I, Doronzo G, Mattiello L, De Salve
A, Trovati M and Anfossi G: The activity of constitutive nitric
oxide synthase is increased by the pathway cAMP/cAMP-activated
protein kinase in human platelets. New insights into the
antiaggregating effects of cAMP-elevating agents. Thromb Res.
114:265–273. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kwiatkowski AV, Gertler FB and Loureiro
JJ: Function and regulation of Ena/VASP proteins. Trends Cell Biol.
13:386–392. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tao Y, Gu YJ, Cao ZH, et al: Endogenous
cGMP-dependent protein kinase reverses EGF-induced MAPK/ERK signal
transduction through phosphorylation of VASP at Ser239. Oncol Lett.
4:1104–1108. 2012.PubMed/NCBI
|
|
14
|
Wheeler S, Siwak DR, Chai R, et al: Tumor
epidermal growth factor receptor and EGFR PY1068 are independent
prognostic indicators for head and neck squamous cell carcinoma.
Clin Cancer Res. 18:2278–2289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yamazaki T, Zaal K, Hailey D, Presley J,
Lippincott-Schwartz J and Samelson LE: Role of Grb2 in
EGF-stimulated EGFR internalization. J Cell Sci. 115:1791–1802.
2002.PubMed/NCBI
|
|
16
|
Rojas M, Yao S and Lin YZ: Controlling
epidermal growth factor (EGF)-stimulated Ras activation in intact
cells by a cell-permeable peptide mimicking phosphorylated EGF
receptor. J Biol Chem. 271:27456–27461. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saito T, Okada S, Ohshima K, et al:
Differential activation of epidermal growth factor (EGF) receptor
downstream signaling pathways by betacellulin and EGF.
Endocrinology. 145:4232–4243. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kolch W: Meaningful relationships: the
regulation of the Ras/Raf/MEK/ERK pathway by protein interactions.
Biochem J. 351:289–305. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dumitrescu C, Biondi R, Xia Y, et al:
Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation
with impaired endothelial function ameliorated by BH4. Proc Natl
Acad Sci USA. 104:15081–15086. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Knowles RG and Moncada S: Nitric oxide
synthases in mammals. Biochem J. 298:249–258. 1994.PubMed/NCBI
|
|
21
|
Koesling D, Russwurm M, Mergia E,
Mullershausen F and Friebe A: Nitric oxide-sensitive guanylyl
cyclase: structure and regulation. Neurochem Int. 45:813–819. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bruckdorfer R: The basics about nitric
oxide. Mol Aspects Med. 26:3–31. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Madhusoodanan KS and Murad F: NO-cGMP
signaling and regenerative medicine involving stem cells. Neurochem
Res. 32:681–694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Russo I, Viretto M, Doronzo G, et al: A
short-term incubation with high glucose impairs VASP
phosphorylation at serine 239 in response to the nitric oxide/cGMP
pathway in vascular smooth muscle cells: role of oxidative stress.
Biomed Res Int. 2014:3289592014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lambrechts A, Kwiatkowski AV, Lanier LM,
et al: cAMP-dependent protein kinase phosphorylation of EVL, a
Mena/VASP relative, regulates its interaction with actin and SH3
domains. J Biol Chem. 275:36143–36151. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Butt E, Abel K, Krieger M, et al: cAMP-
and cGMP-dependent protein kinase phosphorylation sites of the
focal adhesion vasodilator-stimulated phosphoprotein (VASP) in
vitro and in intact human platelets. J Biol Chem. 269:14509–14517.
1994.PubMed/NCBI
|
|
27
|
Tao Y, Chen YC, Sang JR and Xu WR:
Phosphorylation of vasodilator stimulated phosphoprotein is
correlated with cell cycle progression in HeLa cells. Mol Med Rep.
3:657–662. 2010.PubMed/NCBI
|
|
28
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: a family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sang JR, Chen YC and Tao Y: Nitric oxide
inhibits gastric cancer cell growth through the modulation of the
Akt pathway. Mol Med Rep. 4:1163–1167. 2011.PubMed/NCBI
|