Introduction
The autosomal dominant hereditary disorder von
Hippel-Lindau (VHL) disease is caused by a germline mutation in the
VHL gene. VHL is characterized by abundantly vascularized tumors in
multiple organs; such VHL tumors may include hemangioblastoma of
the retina and central nervous system (CNS), renal cell carcinoma
(RCC), pancreatic tumors, pheochromocytoma, endolymphatic sac
tumors, epididymal cystadenoma as well as cysts and cystadenoma in
the kidney, pancreas, epididymis and broad ligament (1). The incidence of VHL disease has been
estimated to be 1 in 36,000 live births in the Caucasian
population, with gradually more apparent clinical manifestations
occurring between ~18 and 30 years of age (2). Clinical diagnostic criteria include a
positive family history and at least one typical VHL tumor. For
isolated cases, at least two typical manifestations, including one
hemangioblastoma, are required for diagnosis (3). Cases of sporadic VHL disease are rare.
Approximately 20% of VHL patients demonstrate negative family
history, with disease resulting from a de novo mutation
(4). The VHL gene has been mapped to
chromosome 3 (3p25–26) (5).
Identification of pathogenic germline VHL mutations has been
reported to provide a reliable method for the confirmation of
diagnosis (6). The current study
reports a case of de novo VHL disease in a male patient with
advanced bilateral multifocal RCC involvement. Written informed
consent was obtained from the patient. The relevant literature is
also reviewed.
Case report
History and examination
A 33-year-old man was admitted to Zhongda Hospital,
Southeast University (Nanjing, China) with a two-month history of
progressive right flank pain, accompanied by a one-month history of
recurrent painless gross hematuria. The patient's previous medical
history included urological surgery at the age of 23 years due to
an epididymal cyst and an ophthalmologic surgery at 26 years of age
for retinal detachment resulting from bilateral retinal
angiomatosis, which was performed at Shuyang People's Hospital
(Suqian, China). At the time of the present study, the patient had
two children, a son and a daughter, who were in good health; his
father, aged 65 years, and his mother, aged 62 years, were also in
good health. The patient's two brothers, aged 40 and 37 years old,
with one child each were all are in good health.
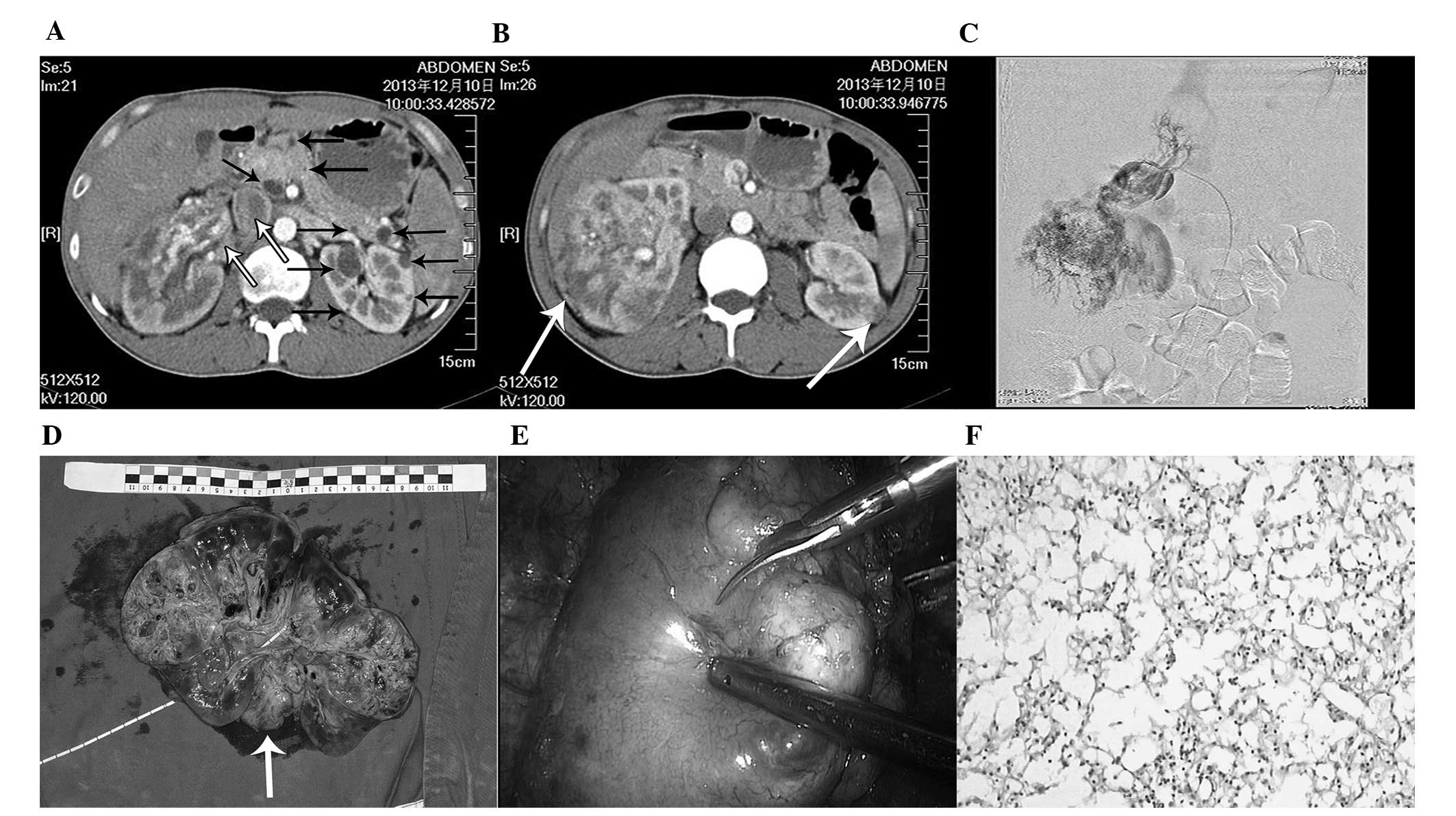
Contrast-enhanced computed tomography (CT) scanning
and angiography were used to detect the presence of quasi-circular
enhancing lesions in the middle of the left kidney and large
irregular enhancing lesions in the right kidney, extending into the
vena cava, that formed an embolus (Fig.
1A–C). In addition, multiple cysts in the pancreas and kidney
were detected radiologically (Fig.
1A).
Surgery and post-surgical course
The patient underwent a radical nephrectomy with an
embolectomy of the right kidney for treatment of
T3b-stage RCC (Fig. 1D).
After three months, he successfully underwent laparoscopic
nephron-sparing surgery (NSS) of the left kidney (Fig. 1E). Histological analysis of the renal
tissue revealed clear cell carcinomas (Fig. 1F). Sunitinib was administered (50 mg
every day for four weeks) following histological diagnosis and
during follow-up.
Genetic study
Peripheral blood samples from nine genetic relatives
of the patient were subjected to VHL mutation analysis. Genomic DNA
was isolated from peripheral blood leukocytes using the CW0541
Blood Gen Midi kit (CW Biotech Co., Ltd., Beijing, China). The
entire coding sequence of the VHL gene, including exon/intron
boundaries, was amplified by polymerase chain reaction (PCR) using
primers (7) manufactured by Generay
Biotech Co., Ltd. (Shanghai, China). The PCR products were directly
sequenced by Nanjing Springen Biotech Co., Ltd. (Nanjing, China)
using the Eppendorf 5331 MasterCycler Gradient Thermal Cycler
(Eppendorf, Hamburg, Germany) under the following conditions:
Denaturation at 95°C for 10 min, followed by 35 cycles of 95°C for
30 sec, 56°C for 30 sec and 72°C for 30 sec, and a final extension
step at 72°C for 10 min. The mutations were confirmed by two-way
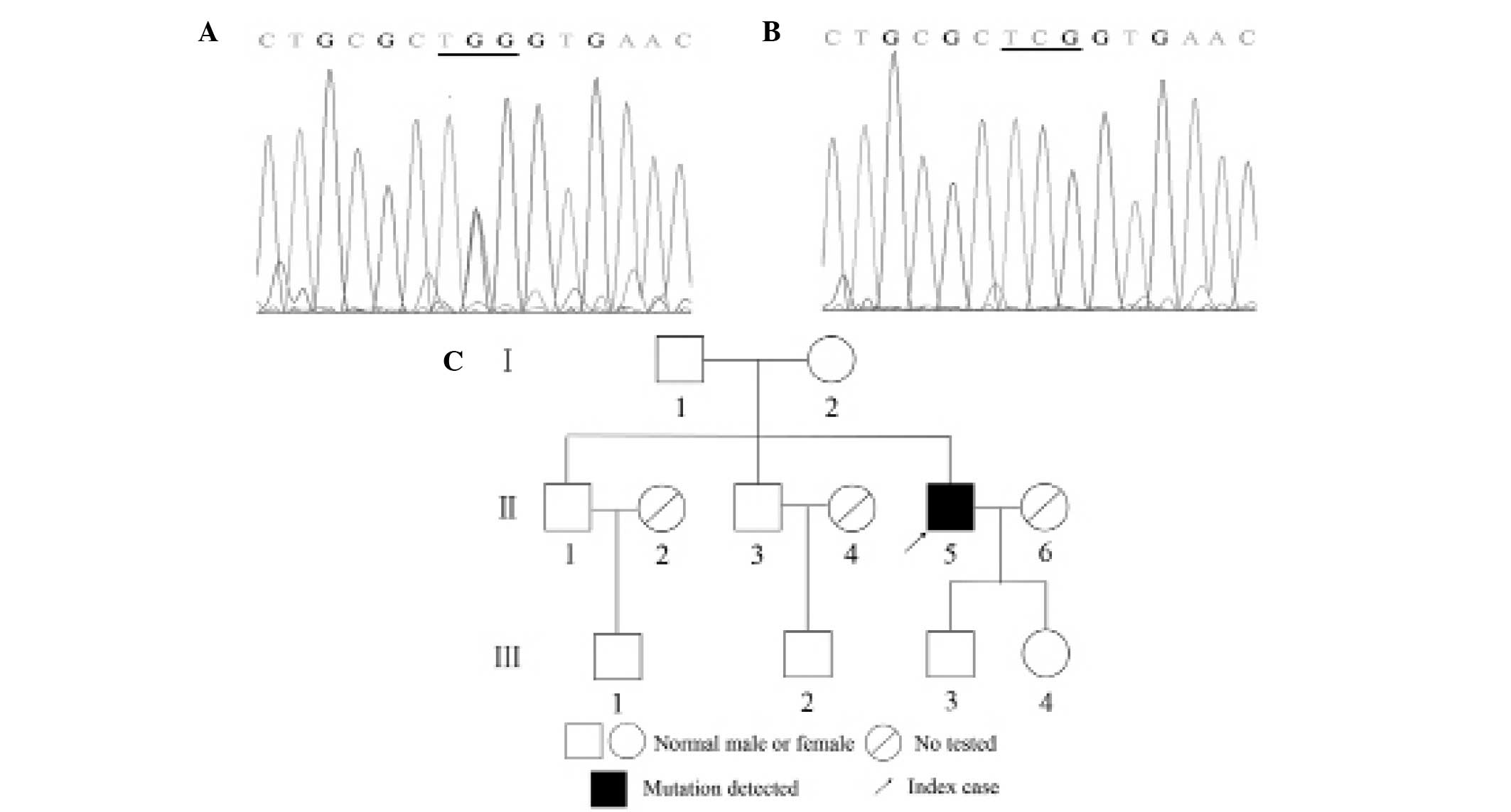
DNA strand sequencing and manually compared. A germline mutation
was detected in exon 1 of the VHL gene [c.194C>G (p.Ser65Trp)]
in genomic DNA from the peripheral blood sample of the patient,
while no mutation was detected in the peripheral blood of the
patient's relatives (Fig. 2).
Discussion
In adults, RCC accounts for ~3% of all cancers and
~85% of all primary malignant kidney tumors (8). In addition, ~5% of RCCs have a
hereditary basis, of which VHL is the most common cause.
Furthermore, ~75% of RCCs have clear cell histology, which is
hereditarily caused by VHL (9). RCC
and cysts in VHL disease are characterized by multicentricity and
bilateral location in >75% of patients (10,11). The
mean age at presentation is 10–20 years earlier than the age
reported for sporadic renal disease (12). Transition from a cyst to a solid
lesion is rare; however, the lining epithelium may be dysplastic or
may have characteristics of carcinoma-in-situ, resulting in
RCC (11). The case reported in the
current study presented with a typical triad of symptoms, including
hematuria, flank pain and flank mass. However, cases with this
‘triad syndrome’ are more advanced and rare; RCC and cysts often
remain asymptomatic for long intervals (13,14). Thus,
serial imaging of the kidneys for monitoring and early diagnosis
has the potential to enhance overall patient outcome. The standard
method for the detection of renal involvement in patients with VHL
is contrast-enhanced abdominal CT imaging (15).
At present, no definitive guidelines are available
with regard to the timing and surgical approach for these
multicentric bilateral tumors. However, the primary aim of
treatment should be cancer control, rather than cure, as well as
the preservation of functional parenchyma in order to avoid the
morbidity associated with renal or adrenal loss. A number of
studies have reported that the majority of RCCs in VHL exhibit a
low pathological grade (16), gradual
growth (17) and do not metastasize
at diameters of <3 cm (18).
Therefore, an individual renal lesion may be maintained under
regular surveillance until it reaches 3 cm in diameter, at which
point NSS is performed to balance the prevention of metastatic
progression against the reduction of the number of interventions
(19). However, studies have revealed
that the majority of patients with VHL-associated RCC eventually
develop local recurrence with the concomitant requirement for
repeated renal surgery (16). Certain
clinicians recommend open NSS as the standard of care for treating
RCC in the setting of VHL disease due to the inability of
laparoscopy to achieve reliable hypothermia, in addition to the
prolonged ischemia and increased complication rates associated with
this approach. However, based on our experiences, the large
incision of the abdominal wall may be problematic, resulting in
muscle injury, the development of hernias and cosmetic damage.
Laparoscopic NSS, performed by experienced surgeons on selected
patients, is an alternative to open surgery (20); the oncological outcome, based on
available series with limited follow-up data, appears to be similar
to the outcome achieved with open NSS (21). Furthermore, an increasing trend
towards the use of percutaneous radiofrequency ablation or
cryoablation for small RCCs has been observed as this method is
less invasive compared with other therapies (22,23).
However, in rare cases with an advanced clinical stage of RCC in
which the kidney cannot be preserved, such as that of the present
study, or probable end-stage renal disease from the reiterated
surgical treatments of localized tumors, total nephrectomy may be
the only option due to a lack of alternative therapies. For
patients who have undergone bilateral nephrectomy, renal
replacement therapy may not be avoided (24,25).
VHL disease occurs due to the inheritance of a
mutated germline copy of the VHL gene from an affected parent. The
wild type copy of the gene is then inactivated by a somatic event.
However, ~20% of patients with VHL develop the disease due to a
de novo mutation. Inactivation of the wild type allele
observed in tumors from VHL patients is consistent with the classic
‘two hit’ hypothesis of tumorigenesis (26,27).
Currently, >400 different mutations have been identified,
including deletion, missense, nonsense, frameshift and insertion
mutations (http://www.umd.be/VHL/W_VHL). In the present study,
the germline mutation c.194C>G of the VHL gene was detected in
the patient; it was predicted that this mutation may induce a
serine-to-tryptophan substitution at amino acid position 65
(p.Ser65Trp) within the VHL protein (pVHL). Serine at amino acid
position 65 is highly conserved among various species and a number
of studies have demonstrated the presence of mutations at this
position (6,28–30).
However, in previous cases involving this mutation, patients were
reported to have a positive family history of VHL disease. In
addition, no relatives of the patient in the current study were
found to carry this mutation or had any clinical indications of VHL
disease. Therefore, for the patient discussed in the present study,
a de novo mutation may have occurred in a germ cell of
either parent or at the embryo stage.
In conclusion, the current study presents a rare
case of a de novo VHL patient with advanced RCC at first
diagnosis; to the best of our knowledge, the patient discussed in
the present study is the first known case of a sporadic de
novo germline mutation of VHL at c.194C>G. Current
understanding of the molecular genetics and pathophysiology of VHL
disease, as well as developments in surgical and target therapies
for RCC have advanced in recent years; however, early detection
through genetic screening and regular clinical surveillance of VHL
disease patients and their families continues to be the predominant
mode of disease management. The enhanced sensitivity of imaging
techniques and advancements in therapeutic techniques, including
radiofrequency ablation and NSS allow for the identification and
management of tumors at earlier stages, thereby attenuating the
morbidity and mortality rates associated with VHL disease.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81370849,
81300472, 81070592 and 81202034), the Natural Science Foundation of
Jiangsu Province (nos. BL2013032 and BK2012336), the Natural
Science Foundation of Nanjing City (no. 201201053), the Natural
Science Foundation of Southeast University (no. 3290002402), the
Science Foundation of Ministry of Education of China (no.
20120092120071) and the Fundamental Research Funds for the Central
Universities and Graduate Innovative Foundation of Jiangsu Province
(no. CXZZ13_0133).
References
|
1
|
Maher ER, Yates JR, Harries R, Benjamin C,
Harris R, Moore AT and Ferguson-Smith MA: Clinical features and
natural history of von Hippel-Lindau disease. Q J Med.
77:1151–1163. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Richard S, Graff J, Lindau J and Resche F:
Von Hippel-Lindau disease. Lancet. 363:1231–1234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaelin WG Jr: Molecular basis of the VHL
hereditary cancer syndrome. Nat Rev Cancer. 2:673–682. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Neumann HP, Riegler P, Huber W, et al: The
challenge of kidney lesions in von Hippel-Lindau disease. Contrib
Nephrol. 136:193–207. 2001.PubMed/NCBI
|
|
5
|
Latif F, Tory K, Gnarra J, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stolle C, Glenn G, Zbar B, et al: Improved
detection of germline mutations in the von Hippel-Lindau disease
tumor suppressor gene. Hum Mutat. 12:417–423. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Geng W, Zhao Y, et al: Clinical
and mutation analysis of four Chinese families with von
Hippel-Lindau disease. Clin Transl Oncol. 15:391–397. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang L, Xu B, Chen S, et al: The complex
roles of microRNAs in the metastasis of renal cell carcinoma. J
Nanosci Nanotechnol. 13:3195–3203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bradley S, Dumas N, Ludman M and Wood L:
Hereditary renal cell carcinoma associated with von Hippel-Lindau
disease: a description of a Nova Scotia cohort. Can Urol Assoc J.
3:32–36. 2009.PubMed/NCBI
|
|
10
|
Malek RS, Omess PJ, Benson RC Jr and
Zincke H: Renal cell carcinoma in von Hippel-Lindau syndrome. Am J
Med. 82:236–238. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choyke PL, Glenn GM, Walther MM, et al:
The natural history of renal lesions in von Hippel-Lindau disease:
A serial CT study in 28 patients. AJR Am J Roentgenol.
159:1229–1234. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choyke PL, Glenn GM, Walther MM, Patronas
NJ, Linehan WM and Zbar B: von Hippel-Lindau disease: Genetic,
clinical and imaging features. Radiology. 194:629–642. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schips L, Lipsky K, Zigeuner R, Salfellner
M, Winkler S, Langner C, Rehak P, Pummer K and Hubmer G: Impact of
tumor-associated symptoms on the prognosis of patients with renal
cell carcinoma: A single-center experience of 683 patients.
Urology. 62:1024–1028. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ares Valdés Y: Correlation between
symptoms and survival in patients with renal cell carcinoma. Arch
Esp Urol. 62:201–206. 2009.(In Spanish). PubMed/NCBI
|
|
15
|
Lonser RR, Glenn GM, Walther M, Chew EY,
Libutti SK, Linehan WM and Oldfield EH: von Hippel-Lindau disease.
Lancet. 361:2059–2067. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Steinbach F, Novick AC, Zincke H, et al:
Treatment of renal cell carcinoma in von Hippel-Lindau disease: A
multicenter study. J Urol. 153:1812–1816. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Neumann HP, Bender BU, Berger DP, et al:
Prevalence, morphology and biology of renal cell carcinoma in von
Hippel-Lindau disease compared to sporadic renal cell carcinoma. J
Urol. 160:1248–1254. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duffey BG, Choyke PL, Glenn G, Grubb RL,
Venzon D, Linehan WM and Walther MM: The relationship between renal
tumor size and metastases in patients with von Hippel-Lindau
disease. J Urol. 172:63–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Herring JC, Enquist EG, Chernoff A,
Linehan WM, Choyke PL and Walther MM: Parenchymal sparing surgery
in patients with hereditary renal cell carcinoma: 10-year
experience. J Urol. 165:777–781. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gill IS, Kavoussi LR, Lane BR, et al:
Comparison of 1,800 laparoscopic and open partial nephrectomies for
single renal tumors. J Urol. 178:41–46. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ljungberg B, Cowan NC, Hanbury DC, et al:
EAU guidelines on renal cell carcinoma: The 2010 update. Eur Urol.
58:398–406. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pavlovich CP, Walther M, Choyke PL,
Pautler SE, Chang R, Linehan WM and Wood BJ: Percutaneous radio
frequency ablation of small renal tumors: Initial results. J Urol.
167:10–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gill IS, Remer EM, Hasan WA, et al: Renal
cryoablation: Outcome at 3 years. J Urol. 173:1903–1907. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goldfarb DA: Nephron-sparing surgery and
renal transplantation in patients with renal cell carcinoma and von
Hippel-Lindau. J Intern Med. 243:563–567. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goldfarb DA, Neumann HP, Penn I and Novick
AC: Results of renal transplantation in patients with renal cell
carcinoma and von Hippel-Lindau disease. Transplantation.
64:1726–1729. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Knudson AG Jr: Genetics of human cancer. J
Cell Physiol. 129 (Suppl 4):7–11. 1986. View Article : Google Scholar
|
|
27
|
Neumann HP and Zbar B: Renal cysts, renal
cancer and von Hippel-Lindau disease. Kidney Int. 51:16–26. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen F, Kishida T, Yao M, et al: Germline
mutations in the von Hippel-Lindau disease tumor suppressor gene:
Correlations with phenotype. Hum Mutat. 5:66–75. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Crossey PA, Richards FM, Foster K, et al:
Identification of intragenic mutations in the von Hippel-Lindau
disease tumour suppressor gene and correlation with disease
phenotype. Hum Mol Genet. 3:1303–1308. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maher ER, Webster AR, Richards FM, Green
JS, Crossey PA, Payne SJ and Moore AT: Phenotypic expression in von
Hippel-Lindau disease: Correlations with germline VHL gene
mutations. J Med Genet. 33:328–332. 1996. View Article : Google Scholar : PubMed/NCBI
|