Introduction
Intracranial atypical teratoid/rhabdoid tumor
(AT/RT) is a rare malignant embryonic neoplasm that usually occurs
in children aged <3 years and accounts for a total incidence of
1–2% of all brain tumors in children and >10% of central nervous
system tumors in infants (1). The
clinical course is dismal, with a median survival time from
diagnosis to mortality that spans only a few months (2). The tumor is typically associated with a
chromosome 22q11.2 mutation or deletion, leading to the loss of
nuclear expression of integrase interactor 1; in adults, the
clinical presentation varies with tumor location (1). Hereditary multiple exostoses (EXT) is
the most common benign bone tumor and is an autosomal dominant
disorder, which is characterized by the formation of
cartilage-capped bone projections (exostoses) localized mainly in
the juxta-epiphyseal region of the long bones. EXT is associated
with two loci, 8q24.1 (EXT1) and 11p11-p13 (EXT2) (3,4). The
condition is characterized by multiple osteochondromas and is
usually painless. The present study reports an adult case of
intracranial AT/RT with a history of EXT and investigates the
possible association between the two tumors. Written informed
consent was obtained from the patient in accordance with the
Declaration of Helsinki. The Ethics Committee of the Xiangya
Hospital of Central South University (Changsha, Hunan, China)
approved all experiments described in the study.
Case report
On October 11 2012, an 18-year-old male was first
admitted to the Xiangya Hospital of Central South University
following one month of progressive projectile vomiting. At this
time, the patient did not complain of a headache or stomach
discomfort. Brain computed tomography (CT) scans that had been
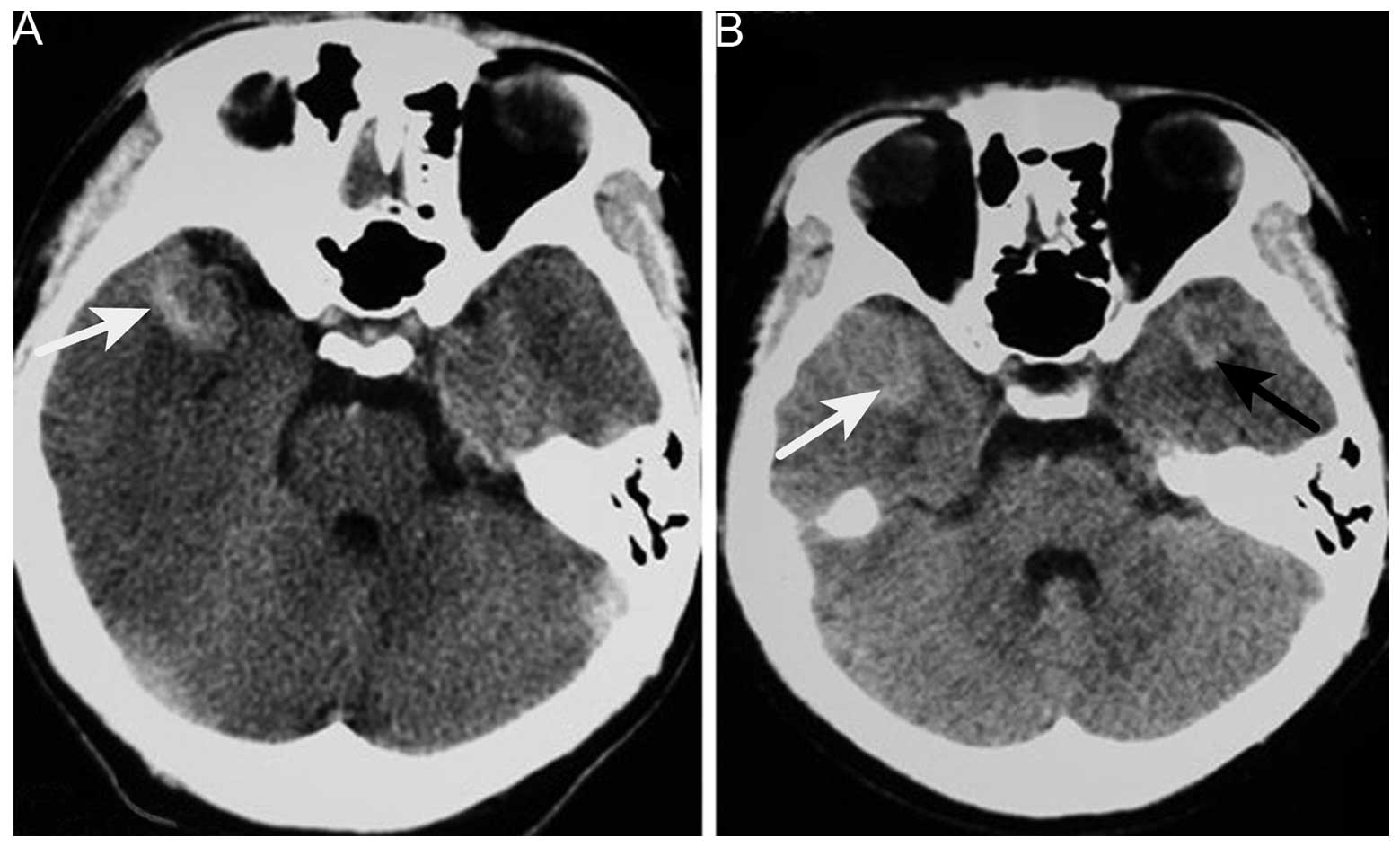
obtained a few days after the symptoms arose revealed a
hyperintense lesion on the right side of the temporal lobe; which
was suspected to be an intracranial hemorrhage at the time.
However, a month of corresponding therapy did not relieve the
vomiting. The patient's past medical history included EXT detected
at 10 years old, and the current examination noted two protuberant
bony deformities over the right scapula and distal humerus. Upon
admission, the neurological examination results were normal.
Hematological and biochemical examination, and cerebrospinal fluid
routine testing were normal; the opening pressure was 100 mm
H2O (normal range, 80–180 mm H2O). Repeat CT
of the brain revealed no changes in the CT value of the lesion
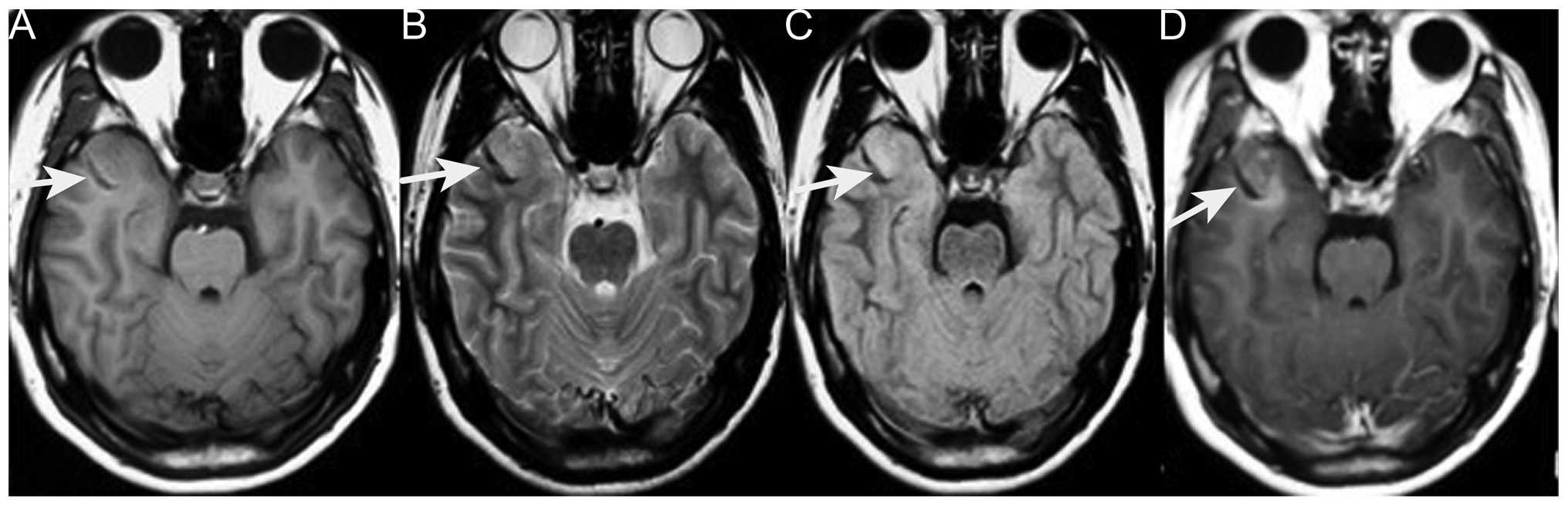
(Fig. 1). Brain magnetic resonance
imaging (MRI) revealed an abnormally low signal intensity on T1-
and T2-weighted images and a high signal intensity on
gadolinium-enhancing T1 in the right lateral fissure and adjacent
temporal region (Fig. 2). Based on
these results, the patient was diagnosed with an intracranial
tumor. Surgery was suggested for removing the mass, which the
patient refused, only accepting carbamazepine (0.1 g, 3 times/day,
October 17–23, 2012) and ondansetron (8 mg, 2 times/day, October
12–16, 2012; 8 mg, 4 times/day, October 17–20, 2012) therapy. Upon
leaving the hospital, the vomiting had been alleviated.
However, 6 months later, the patient was admitted
again due to worsening vomiting and new symptoms of bilateral
blurred vision, and severe neck and waist pain. A neurological
examination revealed sixth cranial nerve palsy and bilateral
papilledema. The lumbar puncture had an opening pressure of >400
mm H2O. Cerebrospinal fluid testing revealed nothing
abnormal and no cancer cells. Brain CT revealed a new hyperintense
lesion in the left temporal region (Fig.
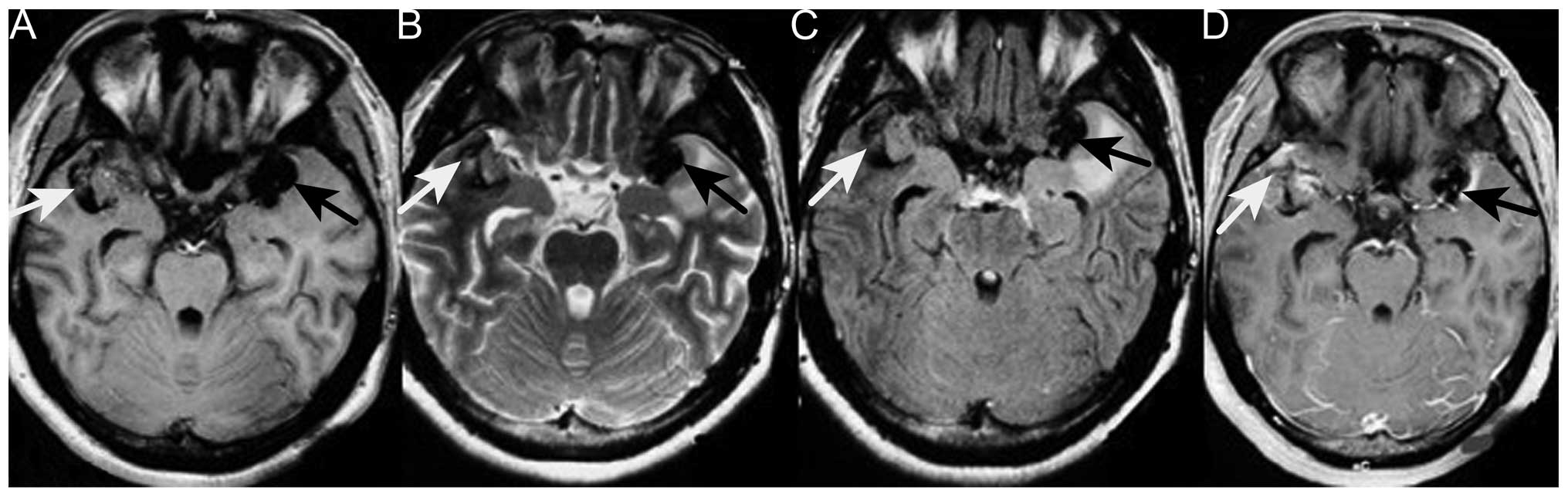
1). Repeat MRI revealed two enhancing masses, with a low
intensity signal on T1-weighted imaging and a heterogeneous signal
on T2-weighted imaging, which had progressed more quickly compared
with the lesion detected 6 months earlier. Moderate peripheral
edema was also observed (Fig. 3).
Digital subtraction angiography disclosed diffuse poor filling of
all venous sinuses during the venous phase. Surgery was performed,
but the patient's condition precluded the use of radiotherapy or
chemotherapy. CT angiography revealed that the tumors were 2.9×1.9
cm (left temporal region) and 2.2×1.9 cm (right temporal region);
surgical microscopy revealed surrounding strong angiogenic
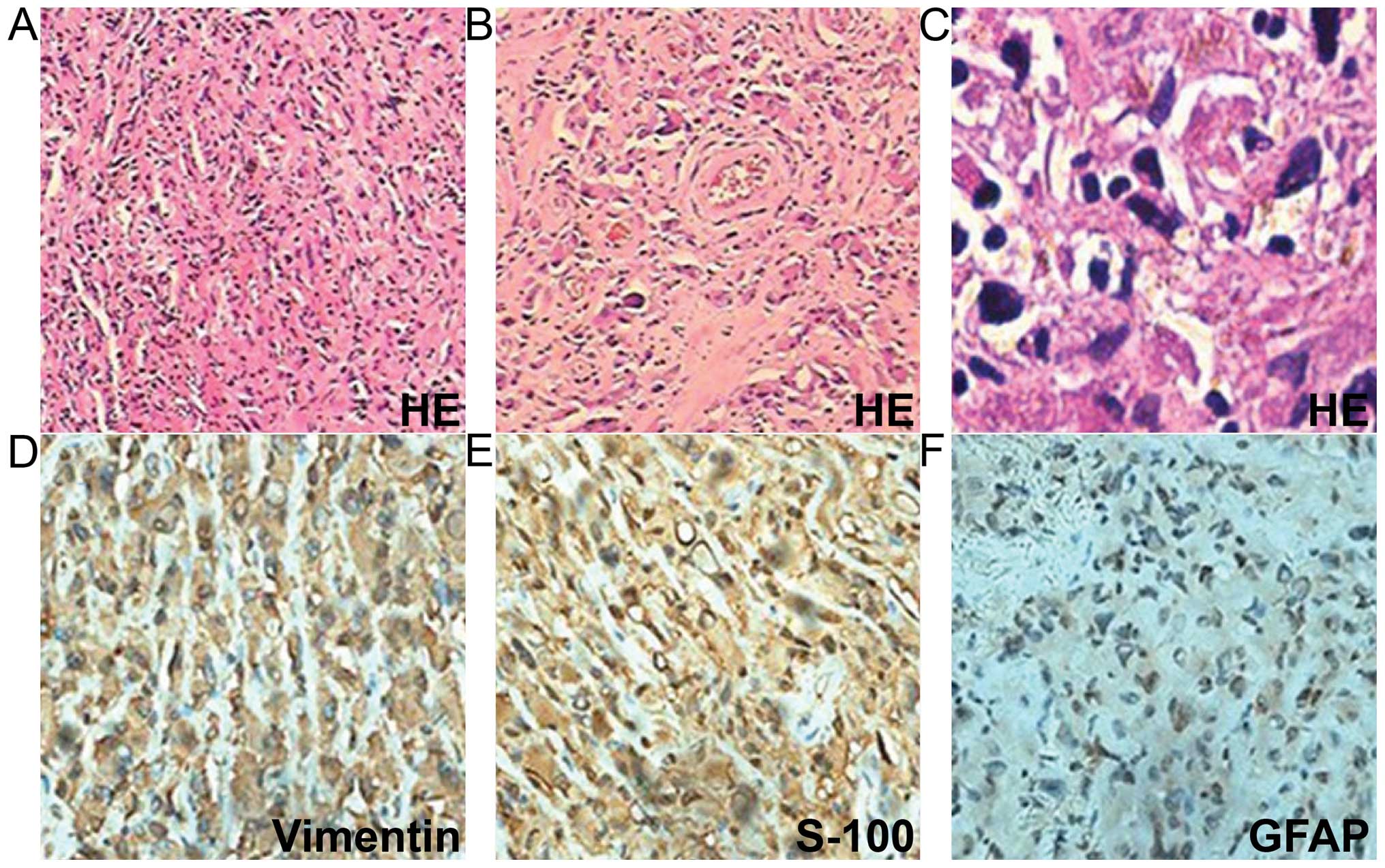
adhesion. The tumor pathology revealed sheets of monotonous medium-
to large-sized neoplastic cells, with abundant eosinophilic
cytoplasm and clear eccentric nucleus with prominent nucleoli. The
tumors were positive for vimentin, glial fibrillary acidic protein
and S-100 (Fig. 4). The Ki-67
labeling index was >2%, and the tumors were negative for cluster
of differentiation (CD)138, CD38, pan-cytokeratin, desmin, muscle
cell actin (HHF35), melanoma marker (HMB45), leukocyte common
antigen, melan-A and myogenin. The diagnosis of intracranial AT/RT
was verified, however, the patient succumbed to pulmonary infection
at 14 days post-surgery.
Discussion
The case reported in the present study is of a rare
adult supratentorial AT/RT, associated with the chief complaint of
vomiting and nausea. The patient's symptoms and physical
examination results cannot be fully attributed to the mass itself
and are more associated with cranial hypertension. To the best of
our knowledge, this is the first description of an AT/RT tumor of
the brain and comorbidity with EXT. Upon first admission, our
primary consideration was intracranial chondrosarcoma. CT and MRI
for intracranial chondrosarcoma also often disclose a mass lesion
that includes areas of calcification and heterogeneous contrast
enhancement. Malignant transformation of exostoses into
chondrosarcoma occurs in 5% of cases (5). Brain metastasis is exceptionally rare,
with few documented cases; lung and breast cancer are the primary
sources of brain metastasis, together accounting for nearly
two-thirds of total cases (6).
Suspicion of malignant transformation is indicated by growth of the
tumor following puberty, the presence of pain, a >1-cm thick
cartilaginous cap in adults, or hints from radiography, bone
scintigraphy (7), and enhanced CT or
MRI. In the present case, the lesion did not increase in size and
bone scintigraphy did not reveal focally increased radiotracer
accumulation. There was no enhancement of the scapula tumor and no
cardiopulmonary neoplasm on enhanced chest CT. With the exception
of metastatic chondrosarcoma, intracranial primary chondrosarcoma
mostly originates from skull base synchondroses and tends to grow
extradurally. These neoplasms include classic, myxoid and
mesenchymal chondrosarcoma (8). The
peak incidence of classic chondrosarcoma is in patients in their
sixth decade, where the tumor typically arises in the skull base.
Mesenchymal chondrosarcoma tends to occur in the second and third
decades of life, and is typically supratentorial, but most often
located in the frontoparietal region and attached to the meninges,
particularly the falx cerebri. The tumor has a biphasic pattern of
undifferentiated mesenchymal cells and well-differentiated
cartilage, and the typical transition between the two states is
clear. Myxoid chondrosarcoma is the rarest form, and reported
locations include the petrous bone, posterior fossa and falx
cerebri. Histologically, the tumor is characterized by prominent
mucinous supporting stroma. Intracranial primary chondrosarcoma has
a tendency to locate in the skull base or the falx cerebri.
However, the tumor in the present case was near neither the skull
nor the falx cerebri. The differential diagnosis included
oligodendroglioma, germinoma and other tumors containing
calcification.
The present case was an uncommon example of an AT/RT
tumor with typical histological features, but presenting in the
supratentorial compartment of an adult. The most notable aspect was
that the patient had a history of EXT. From the literature, we
infer that the two tumors may be genetically correlated. Several
studies reported that AT/RT was associated with insulin-like growth
factor II (IGF2) and IGF receptor type 1, and that
autocrine/paracrine stimulation of cell growth by IGF2, which is on
chromosome 11p15 and near EXT2, may be a mechanism involved in
AT/RT tumorigenesis (9–12). Therefore, AT/RT can theoretically
occur together with skeletal dysplasia. However, given the scarcity
of cases and limitations of the experimental infrastructure,
conclusions cannot be derived at present. Instead, the current case
report is presented to improve the approach to diagnosing AT/RT and
aid more patients.
References
|
1
|
Shonka NA, Armstrong TS, Prabhu SS,
Childress A, Choi S, Langford LA and Gilbert MR: Atypical
teratoid/rhabdoid tumors in adults: a case report and
treatment-focused review. J Clin Med Res. 3:85–92. 2011.PubMed/NCBI
|
|
2
|
Lafay-Cousin L, Hawkins C, Carret AS,
Johnston D, Zelcer S, Wilson B, Jabado N, Scheinemann K, Eisenstat
D, Fryer C, et al: Central nervous system atypical teratoid
rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium
experience. Eur J Cancer. 48:353–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Solomon L: Hereditary multiple exostosis.
J Bone Joint Surg. 45:292–304. 1963.
|
|
4
|
Li Y1, Wang D, Wang W, Wang J, Li H, Wang
J, Wang X and Fu Q: Identification of four novel EXT1 and EXT2
mutations in five Chinese pedigrees with hereditary multiple
exostoses. Genet Test Mol Biomarkers. 13:825–830. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Altay M, Bayrakci K, Yildiz Y, Erekul S
and Saglik Y: Secondary chondrosarcoma in cartilage bone tumors:
Report of 32 patients. J Orthop Sci. 12:415–423. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valiente M, Obenauf AC, Jin X, Chen Q,
Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E and Massagué
J: Serpins promote cancer cell survival and vascular co-option in
brain metastasis. Cell. 156:1002–1016. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sánchez-Rodríguez V, Medina-Romero F,
Gómez Rodríguez-Bethencourt MÁ, González Díaz MA, González Soto MJ
and Alarcó Hernández R: Value of the bone scintigraphy in multiple
osteochrondromatosis with sarcomatous degeneration. Rev Esp Med
Nucl Imagen Mol. 31:270–274. 2012.PubMed/NCBI
|
|
8
|
Im SH, Kim DG, Park IA and Chi JG: Primary
intracranial myxoid chondrosarcoma: Report of a case and review of
the literature. J Korean Med Sci. 18:301–307. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogino S, Cohen ML and Abdul-Karim FW:
Atypical teratoid/rhabdoid tumor of the CNS: Cytopathology and
immunohistochemistry of insulin-like growth factor-II, insulin-like
growth factor receptor type 1, cathepsin D, and Ki-67. Mod Pathol.
12:379–385. 1999.PubMed/NCBI
|
|
10
|
Tessema M, Länger F, Bock O, Seltsam A,
Metzig K, Hasemeier B, Kreipe H and Lehmann U: Down-regulation of
the IGF-2/H19 locus during normal and malignant hematopoiesis is
independent of the imprinting pattern. Int J Oncol. 26:499–507.
2005.PubMed/NCBI
|
|
11
|
Lopez-Gines C, Cerda-Nicolas M, Kepes J,
Donat J, Gil-Benso R and Llombart-Bosch A: Complex rearrangement of
chromosomes 6 and 11 as the sole anomaly in atypical
teratoid/rhabdoid tumors of the central nervous system. Cancer
Genet Cytogenet. 122:149–152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Narendran A, Coppes L, Jayanthan A, Coppes
M, Teja B, Bernoux D, George D and Strother D: Establishment of
atypical-teratoid/rhabdoid tumor (AT/RT) cell cultures from
disseminated CSF cells: A model to elucidate biology and potential
targeted therapeutics. J Neurooncol. 90:171–180. 2008. View Article : Google Scholar : PubMed/NCBI
|