Introduction
Chronic neutrophilic leukemia (CNL) is a rare
myeloproliferative neoplasm (MPN) that is primarily characterized
by leukocytosis, but is often lacking in distinct clinical,
laboratory and molecular features (1). The majority of CNL cases are fatal, most
often due to severe cerebral hemorrhage, with a median survival
time of <2 years (2). Management
is typically symptomatic, however, allogeneic transplantation in
younger patients may represent a curative treatment strategy. The
optimal therapeutic regime for CNL remains uncertain (3). Until recently, the molecular
pathogenesis of CNL was unknown; therefore, diagnoses were based on
morphological analysis, clinical criteria and the exclusion of
known genetic entities, such as mutations of the breakpoint cluster
region (BCR)/ABL proto-oncogene 1 (ABL1) gene
transcript or Janus kinase 2 (JAK2).
Colony-stimulating factor 3 (CSF3R) encodes
the receptor for CSF3, a cytokine that controls the generation,
differentiation and function of granulocytes (4,5). Mutations
in CSF3R are associated with severe congenital neutropenia.
Maxson et al (6) recently
demonstrated that CSF3R mutations are associated with CNL
and atypical chronic myeloid leukemia (aCML). In addition, the
following association between CNL and activating CSF3R
mutations was established: Oncogenic CSF3R mutations may be
used as molecular markers of sensitivity to SRC family tyrosine
kinase non-receptor 2 and JAK inhibitors. The aforementioned
studies may facilitate the development of novel therapeutic
strategies for CNL (7). In humans,
ecotropic viral integration site-1 (EVI-1, also known as
MECOM) is located at chromosome 3q26 and, in cases of
hematological malignancy, rearrangements at this locus frequently
lead to increased EVI-1 expression. In addition, overexpression of
EVI-1 occurs with high frequency in leukemia patients who do not
possess chromosome 3q26 abnormalities. Therefore, high EVI-1
expression is an independent negative prognostic indicator for
certain types of cancer, irrespective of the presence of chromosome
3q26 rearrangements (8). SET binding
protein-1 (SETBP1) stabilizes the protein SET, an inhibitor
of the tumor suppressor protein phosphatase 2A (PP2A).
SETBP1-mutated cells express higher levels of SETBP1 and
thus, exhibit lower PP2A activity with higher proliferative rates
compared with their wild-type SETBP1 counterparts (9). Clinically, patients with SETBP1
mutations exhibit a significantly higher number of leukocytes and a
worse prognosis than patients with wild-type SETBP1
(9). A previous study demonstrated
that 4/12 (33%) patients with CNL were found to carry a
SETBP1 mutation. All patients coexpressed the
CSF3RT618I mutation and exhibited a trend towards
reduced survival (10). Therefore,
SETBP1 mutations may be a prognostic indicator for CNL.
The present study reports a patient with
double-mutated CNL exhibiting CSF3RT618I and
SETBP1D868N with associated overexpression of
EVI-1. To the best of our knowledge, this is the first reported
case of CNL with an associated mutation in EVI-1.
Case report
Presentation
On September 4, 2012, a 67-year-old man was admitted
to the First Central Clinical College of Tianjin Medical University
(Tianjin, China) with a number of symptoms, including ecchymosis,
fatigue, and edema and pain in the right leg. Skin ecchymosis had
been present for 4 months and was not associated with bleeding from
any orifice, or with a history of deep bleeding. The patient
exhibited edema and pain in the right leg 10 days prior to
admission that caused difficulty walking. These symptoms were
associated with fatigue, however, there was no evidence to indicate
an infection. The patient had no significant history of recent
cytotoxic, immunosuppressive or growth factor therapy, or exposure
to chemicals. Furthermore, the patient's personal and family
medical history were not relevant to the symptoms exhibited.
Physical examination revealed mild pallor, a small number of
ecchymotic patches on the skin and considerable edema in right leg.
However, hypertrophy of the gums, hepatomegaly, splenomegaly or
enlargement of the peripheral lymph nodes were not observed.
The ethics committee of the First Central Clinical
College of Tianjin Medical University approved the use of
patient-derived cells and the protocols of the present study.
Written informed consent was obtained from the patient's
family.
Clinical investigation
Laboratory investigations performed upon initial
admission revealed a hemoglobin level of 69 g/dl (normal range,
130–175 g/dl), a red blood cell count of 2.41×1012/l
(normal range, 4.31–5.82×1012/l), a white blood cell
count of 49.41×109/l (normal range,
3.50–9.49×109/l) and a platelet count of
94×109/l (normal range, 150–350×109/l). A
peripheral blood film (98.5% neutrophils, 1.30% lymphocytes, 0.01%
monocytes, 0.10% eosinophils and 0.09% basophils) indicated
leukocytosis with an increased number of segmented and band-stage
neutrophils. A small number of myelocytes and an occasional blast
were noted, however, basophilia or eosinophilia were not
observed.
Serum electrolyte levels and liver function test
results were normal, however, lactate dehydrogenase levels were
high at 925.1 U/l (normal range, 135.0–255.0 U/l). A serum assay
indicated elevated ferritin (688.6 ng/ml; normal range, 15.0–200.0
ng/ml), vitamin B12 (1,512 pg/ml; normal range,
210–1,100 pg/ml) and uric acid (567 µmol/l; normal range, 238–506
µmol/l) levels. Furthermore, the patient's peripheral blood
neutrophil alkaline phosphatase (NAP) score was 292.8 (normal
range, 69.9–103.3). However, laboratory investigations did not
detect any monoclonal protein in the serum or urine and coagulation
tests were normal. Bone marrow aspiration and biopsy demonstrated a
notable hypercellular marrow with marked granulocytic
proliferation, predominantly consisting of band-stage and segmented
neutrophils, with no dysplastic changes evident. Erythroid and
megakaryocytic cell levels were depressed, however, the morphology
was normal. In addition, the myeloid:erythroid ratio was 9.8:1, and
there was no increase in the proportion of basophils and
eosinophils. A trephine biopsy of the bone marrow demonstrated
similar results and there was no increase in the number of
reticulin fibres. Systemic examinations using X-rays, computed
tomography scans and analysis of the tumor markers, including
α-fetoprotein, acid phosphatase and prostatic-specific antigen,
demonstrated no evidence of malignancy. Furthermore,
ultrasonography of the abdomen did not reveal any abnormalities and
there appeared to be no detectable underlying disease causing the
leukemoid reaction. Flow cytometry analysis was consistent with
marked myeloid hyperplasia without an increase in the number of
blasts or monocytes.
Conventional cytogenetic analysis revealed a normal
karyotype [46, XY (16 cells)] and an absence of the Philadelphia
(Ph) chromosome. Furthermore, molecular biology investigations
demonstrated negativity for the p210 BCR/ABL1 (e13a2, e14a2), p230
BCR/ABL1 (e19, e20) and p190 BCR/ABL1 (e1a2) fusion proteins, an
absence of the JAK2V617F mutation, and no
mutations in the platelet derived growth factor receptor α/β
polypeptides (PDGFRA/PDGFRB). Additional genetic
analysis was conducted using bone marrow samples, qualitatively
screening 31 leukemia fusion genes by nested polymerase chain
reaction (PCR) using the Whole Blood RNA Extraction Kit (BioChain
Institute, Inc., Newark, CA, USA). The following genes were
included in the screen: MLL (also known as KMT2A)/AFX (also
known as FOXO4), MLL/AF6 (also known as MLLT4),
MLL/ELL, CBFB/MYH11, MLL/AF1P (also known as
EPS15), MLL/AF10 (also known as MLLT10), MLL/AF17
(also known as MLLT6), dupMLL, E2A (also known as
TCF3)/PBX1, E2A/HLF, STIL/TAL1, TEL
(also known as ETV6)/AML1 (also known as RUNX1),
AML1/ETO, AML1/EVI1, TLS (also known as
FUS)/ERG, MLL/AF1Q (also known as MLLT11),
MLL/AF4 (also known as AFF1), MLL/AF9 (also known as
MLLT3), MLL/ENL (also known as MLLT1),
BCR/ABL1, TEL/PDGFRB, DEK/CAN (also known as
NUP214), SET/CAN, PML/RARA, PLZF (also
known as ZBTB16)/RARA, NPM1/ALK, NPM1/MLF1,
NPM1/RARA, TEL/ABL1, HOX11 (also known as
TLX1) and EVI-1. The results of this qualitative analysis
demonstrated that only EVI-1 was expressed in the bone marrow
(Table I). During the clinical
course, karyotyping and expression analysis of the BCR-ABL1
transcripts and EVI-1 gene were repeated three times, with similar
results.
 | Table I.Qualitative and quantitative EVI-1
screening tests. |
Table I.
Qualitative and quantitative EVI-1
screening tests.
| Test | Bone marrow
sample | EVI-1
expression |
| Qualitative
screening of 36 leukemic fusion genes |
|
|
|
September 13, 2012 | Total | Positive |
| Quantitative
screening of EVI-1 expression |
|
|
|
September 25, 2012 | Total | 499.87a (overexpression) |
|
November 5, 2012 | CD34+
cell fraction | 214.82a (overexpression) |
|
November 5, 2012 | CD15+
cell fraction |
34.21a (low expression) |
| May 15,
2012 | Total | 151.27a (overexpression) |
In consideration of the results of the nested PCR
screen, correlation between EVI-1 expression and the pathogenesis
of CNL was investigated by analyzing cell apoptosis, migration and
cell cycle distribution. First, neutrophils were purified from bone
marrow samples by Ficoll-Hypaque gradient centrifugation and
erythrocytes were removed using EasySep™ Red Blood Cell Lysis
Buffer (STEMCELL Technologies, Inc., Vancouver, BC, Canada). After
washing three times, the purified neutrophils were labeled with
fluorescein isothiocyanate (FITC)-cluster of differentiation (CD)15
(BD Biosciences, Franklin Lakes, NJ, USA). The labeled cells were
analyzed using flow cytometry (LSR II; BD Biosciences) and 93.9%
were identified as mature neutrophils.
To investigate apoptosis, the purified neutrophils
were cultured in RPMI 1640 medium with 10% fetal bovine serum.
Cells were collected at specific time points (0, 6, 12, 24, 48, 72
and 96 h), and labeled with Annexin V and propidium iodide (BD
Biosciences). Apoptosis of the patient's neutrophils was measured
using flow cytometry (LSR II; BD Biosciences) and compared with the
apoptosis of neutrophils obtained from healthy donors (control
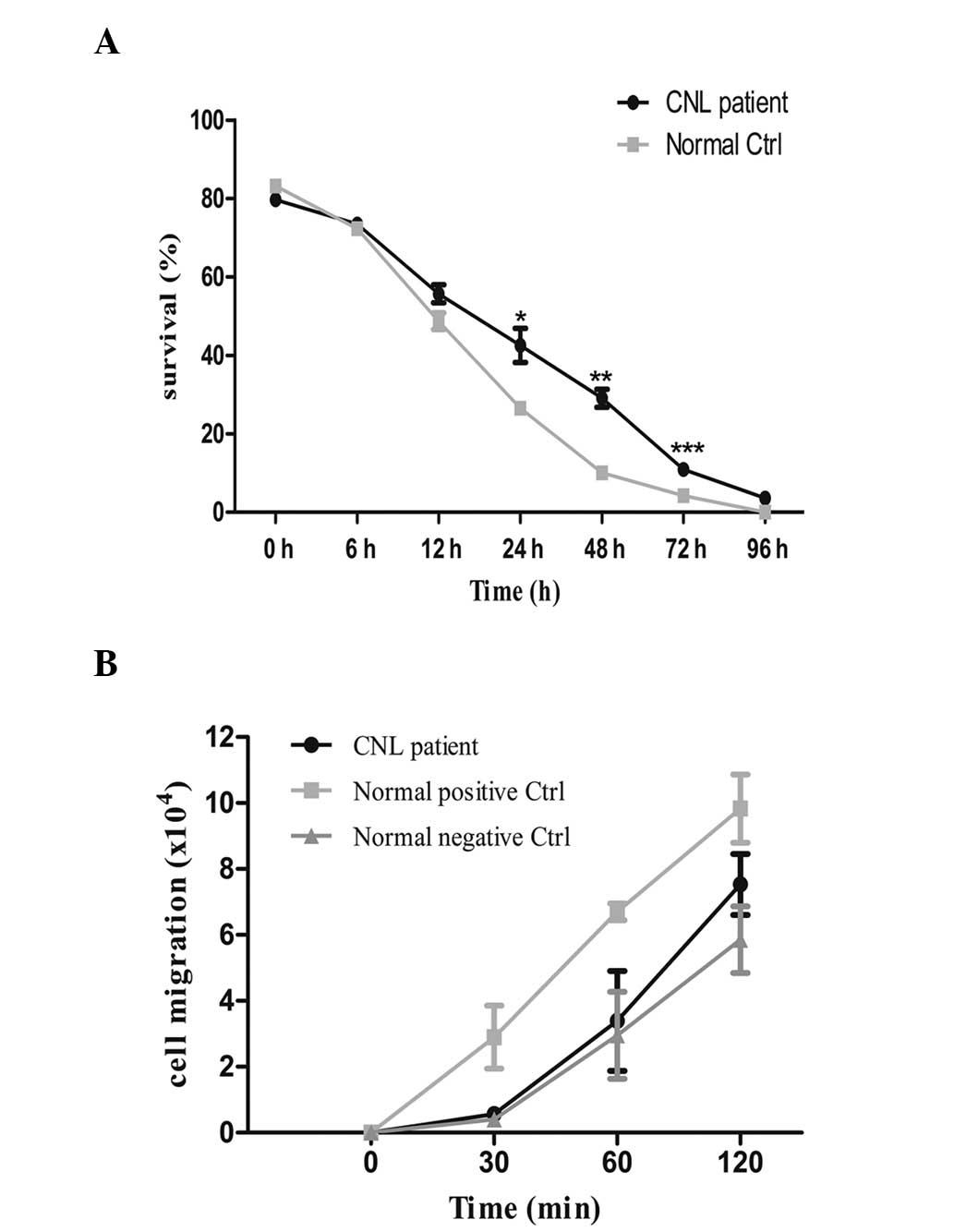
group cells). Flow cytometric analysis revealed that a
significantly higher number of patient neutrophils survived to 72 h
compared with control neutrophils (Fig.
1A; P=0.0010).
To assay migration, the patient-derived purified
neutrophil sample, and the negative and positive control cells were
cultured in Transwell® inserts (Corning, Inc., Corning, NY, USA)
(1×106 cells/well). Subsequently, 1 µM chemotactic
peptide N-formyl-methionyl-leucyl-phenylalanine (fMLP;
Sigma-Aldrich, St. Louis, MO, USA) was added to the lower
compartment. The number of the cells in the lower compartment was
counted at each time point (0, 30, 60 and 120 min). The data
revealed that fMLP-induced cell migration was lower in the
patient-derived neutrophils compared with the positive control
cells incubated with fMLP, but higher than negative control
neutrophils incubated without fMLP. However, the differences were
not statistically significant (Fig.
1B). Cell apoptosis and migration data were analyzed using
Graphpad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA).
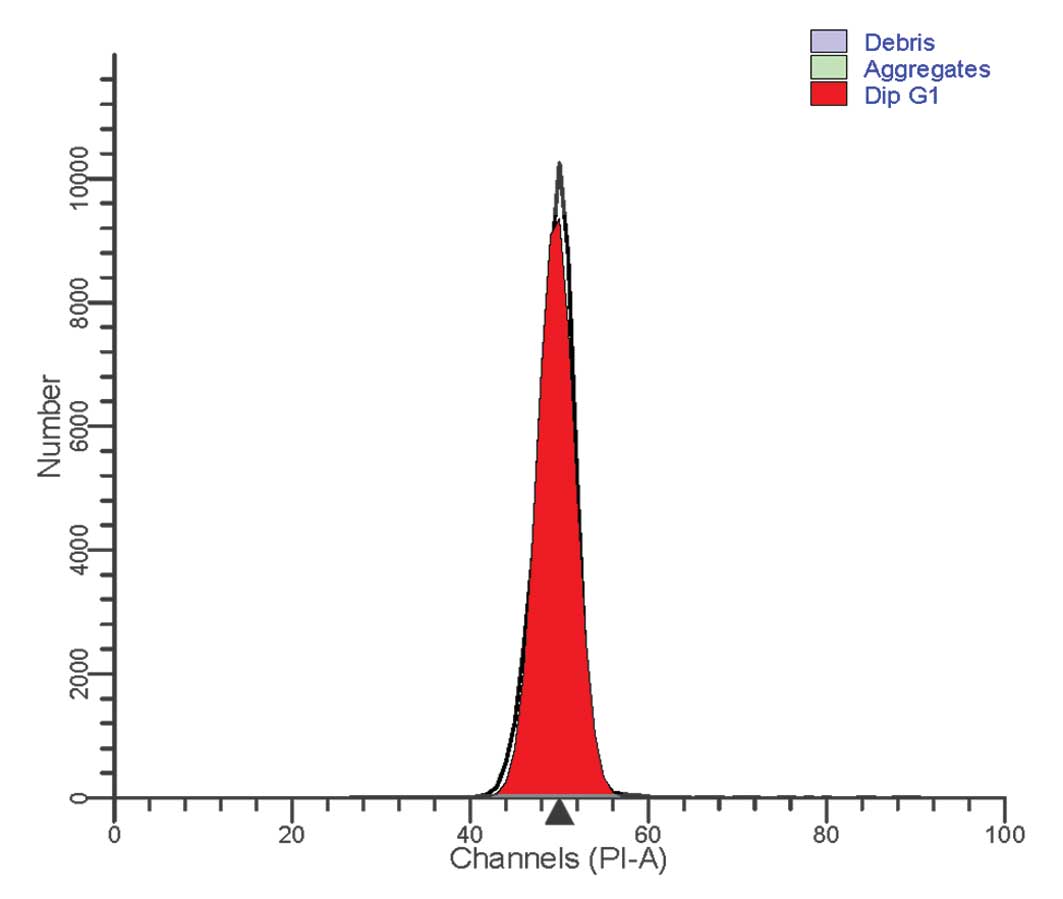
For cell cycle analysis of the patient-derived
neutrophils, a Cycletest™ Plus DNA Reagent kit (BD Biosciences) was
used, according to the manufacturer's instructions. Flow cytometric
analysis of the cell cycle revealed a normal distribution. All
cells were terminally differentiated and no cell division was
observed (Fig. 2).
Following detection of EVI-1 overexpression by a
nested PCR screen of 31 leukemia fusion genes, the expression of
EVI-1 was quantitatively analyzed in September 2012. This was
achieved by sorting the bone marrow samples into CD34+
and CD15+ cell fractions (BD FACSAria™ III;
BD Biosciences), and performing reverse transcription
quantitative-PCR (RT-qPCR) to detect EVI-1 expression at three time
points during the clinical course. Fluorescent Taqman® probes
(Applied Biosystems Life Technologies, Foster City, CA, USA) were
used to detect the expression of EVI-1 and an endogenous reference
gene (ABL1) using the following oligonucleotide primers:
Forward, 5′-GTACTTGAGCCAGCTTCCAACA-3′, and reverse,
5′-CTTCTTGACTAAAGCCCTTGGA-3′ for EVI-1; and forward, 5′-TGG AGA TAA
CAC TCT AAG CAT AAC TAA AGG T-3′, and reverse, 5′-GAT GTA GTT GCT
TGG GAC CCA-3′ for ABL1. Cycle threshold (Ct) values (LightCycler;
Roche Diagnostics, Basel, Switzerland) of bone marrow samples
obtained from healthy volunteers (control) were then used to
calculate the relative quantification of EVI-1 expression, as
follows: ΔCtcontrol = Ctcontrol EVI1 -
Ctcontrol ABL1 and ΔCtpatient =
Ctpatient EVI1 - Ctpatient ABL1.
Subsequently, ΔΔCt = ΔCtpatient - ΔCtcontrol
was calculated and used to determine the fold-change in EVI-1 gene
expression, defined as 2−ΔΔCt.
The 2−ΔΔCt value was <8 in the healthy
control samples. Based on this data, the present study used the
following criteria for categorizing gene expression:
2−ΔΔCt≤8, low EVI-1 expression; 8≤2−ΔΔCt≤32,
middle EVI-1 expression; and 2−ΔΔCt≥32, high EVI-1
expression. Notably, the RT-qPCR data yielded similar results
across samples, particularly for the detection of EVI-1
overexpression in total bone marrow samples and CD34+
cell fractions, however, the CD15+ cell fraction
demonstrated overexpression of EVI-1 (Table I).
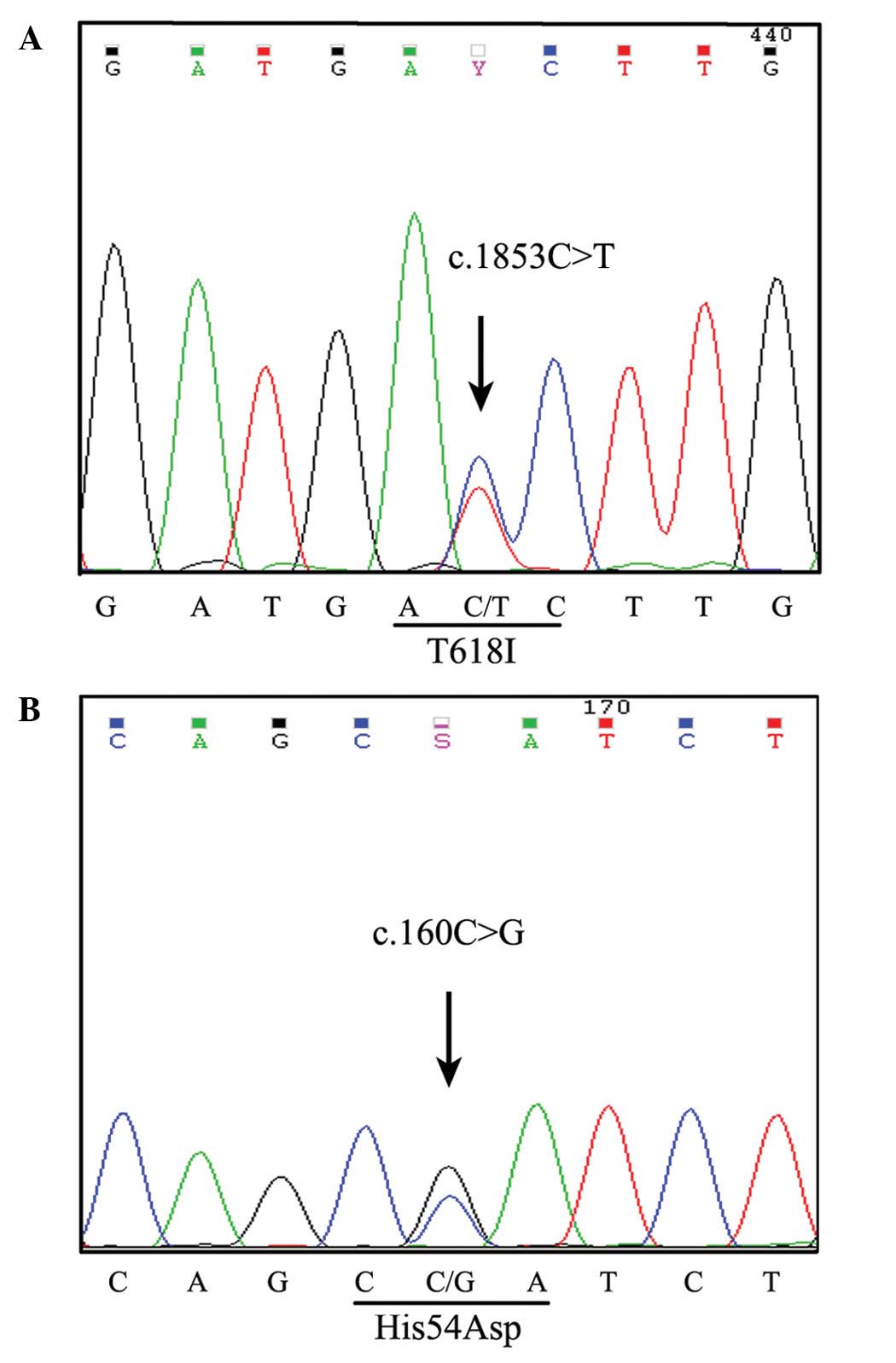
A CSF3R mutation spanning an entire exon and
intron was detected in the CD34+ and CD15+
cell fractions using a PCR-based DNA pyrosequencing method
(6,7).
The p.Thr618Ile membrane proximal mutation was detected on exon 14
of CSF3R (Fig. 3A).
Additionally, a novel mutation site was revealed on exon 4 of the
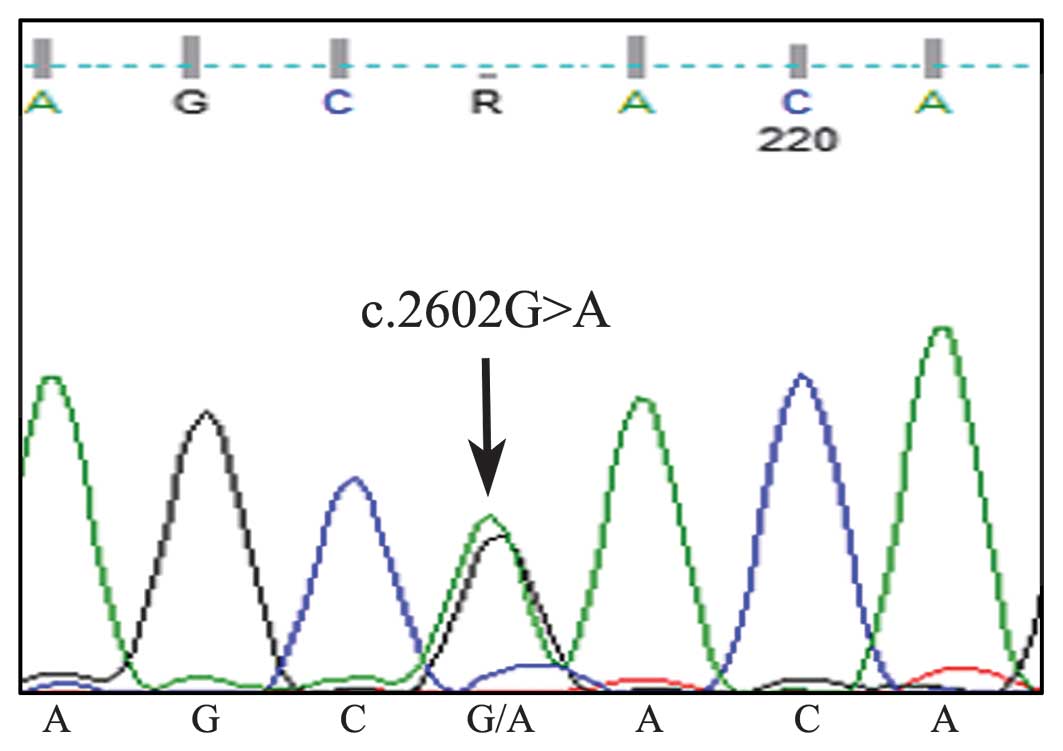
p.His54Asp heterozygous CSF3R mutation (Fig. 3B). Somatic SETBP1 mutational
alteration analysis was performed by PCR amplification followed by
Sanger sequencing for codons located at base pairs 706–917 of exon
3. The results of this analysis indicated that a somatic
heterozygous SETBP1 mutation was encoded at the location
p.Asp868Asn, c.2602C>A (Fig.
4).
Differential diagnosis
The assessment of a patient with atypical
myeloproliferative features and the determination of a correct
diagnosis of CNL may pose a challenge for clinicians. In the
present study, the World Health Organization (WHO) diagnostic
criteria (11) was used to determine
the following differential diagnoses upon admission: CML, aCML, CNL
and reactive leukocytosis. Using the additional data obtained in
the aforementioned laboratory investigations, the patient's initial
differential diagnosis was further restricted to aCML versus CNL.
Utilizing the rigorous diagnostic criteria defined by the WHO, a
direct comparison of the clinical and laboratory features of the
case was performed. The patient met all the criteria for a
diagnosis of CNL, while a diagnosis of aCML was less likely
considering the lack of evidence of overt neutrophilic
dysgranulopoiesis, a virtual absence of circulating immature
myeloid cells and the demonstration of only mild splenomegaly upon
physical examination (Table II).
Thus, a diagnosis of CNL was determined and the patient's clinical
course was followed up for 15 months.
| Table II.WHO diagnostic criteria for CNL and
aCML with corresponding patient clinical data. |
Table II.
WHO diagnostic criteria for CNL and
aCML with corresponding patient clinical data.
|
| WHO diagnostic
criteria | Fulfills
criterion |
|---|
|
|
|
|
|---|
| Patient data | CNL | aCML | CNL | aCML |
|---|
| 25×109/1
WBC, including >80% neutrophils, and no dysgranulopoiesis |
≥25×109/1 WBCs, including
>80% segmented neutrophilsa |
≥13×109/1 WBCs with
dysgranulopoiesis | + | – |
| Hypercellular
marrow with mature cells | Hypercellular
marrowb | Hypercellular
marrowc | + | + |
| No Ph or
BCR/ABL1 fusion gene | No Ph or
BCR/ABL1 fusion gene | No Ph or
BCR/ABL1 fusion gene | + | + |
| No rearrangement of
PDGFRA/PDGFRB | No rearrangement of
PDGFRA/PDGFRB or FGFR1 | No rearrangement of
PDGFRA/PDGFRB | + | + |
| Blood neutrophil
precursors comprising <10% of WBCs | Immature
granulocytes comprising <10% of WBCs | Blood neutrophil
precursors comprising ≥10% of WBCs | + | – |
| No basophilia in
the blood or bone marrow | No basophilia in
the blood or bone marrow | Minimal basophilia
(<2%) in the blood and bone marrow | + | + |
| 1% monocytes | No monocytosis | Minimal monocytosis
(<10%) | + | + |
| <20% blasts in
the blood and marrow | <1% myeloblasts
in the blood and bone marrow | <20% blasts in
the blood and marrow | + | + |
| Mild
hepatosplenomegaly |
Hepatosplenomegaly | No
hematosplenomegaly | + | – |
| No physiologic
cause for neutrophilia | No physiological
cause for neutrophilia | No physiological
cause for neutrophilia | + | + |
| No evidence of PV,
ET or PM | No evidence of PV,
ET or PM | No evidence of PV,
ET or PM | + | + |
| No evidence of MDS
or | No evidence of MDS
or | No evidence of MDS
or | + | + |
| MDS/MPD | MDS/MPD | MDS/MPD |
|
|
Treatment
On September 14, 2012, the patient commenced with a
treatment strategy of hydroxyurea (1,500 mg/day) and subcutaneous
interferon-α (IFN-α; 300 IU/day, three times a week). The patient's
leukocyte count decreased to 7.67×109/l following 3
weeks of treatment (76.8% neutrophils, 12.3% lymphocytes, 9.6%
monocytes, 1.0% eosinophils and 0.3% basophils). As a result of
this, the doses were reduced to 500 mg/day hydroxyurea and 300
IU/day IFN-α, two times a week. The patient's treatment continued
for the next 15 months, with monitoring and control of the
leukocyte count within a range of 15–30×109/l. In
September 2013, ~1 year subsequent to the patient's initial
admission, the patient exhibited bleeding gums, splenomegaly and
lymphadenopathy. The splenomegaly was palpable 4 cm below the
costal margin, and lymph nodes of the cervical and inguinal folds
were enlarged to 1.5 cm. The platelet count persistently decreased
from 90×109/l to 20×109/l during the clinical
course. Adjustment of the treatment dose was difficult due to this
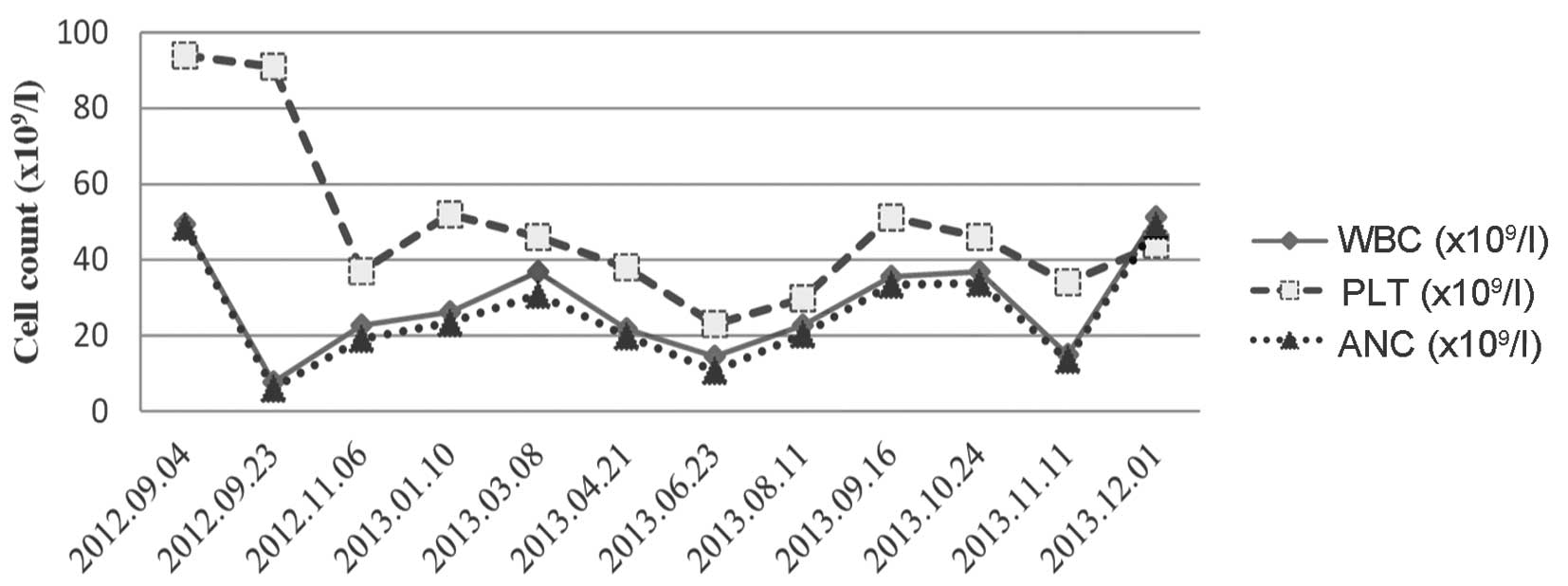
progressive thrombocytopenia (Fig.
5). In December 2013, the leukocyte count fluctuated and
increased to 52×109/l. Later in December 2013, the
patient succumbed to a serious fungal infection of the lung.
Discussion
Following the initial description of CNL in 1920
(12), >150 cases have been
reported in the literature (13).
However, EVI-1 overexpression has not been observed in any of these
cases. CNL is a rare disorder and can be distinguished from CML by
an absence of the Ph chromosome or the BCR/ABL1
fusion gene. In addition, rearrangements in genes encoding
PDGFRA/PDGFRB and fibroblast growth factor receptor 1
are not present. CNL is an MPN characterized by persistent
neutrophilia and splenomegaly, and typically affects elderly
patients of both genders. The majority of patients with CNL have a
poor prognosis, with a mean survival time of 21 months (2,3). However,
Reilly performed a survival analysis study of 33 patients
considered to have CNL and identified an overall median survival
time of 30 months, with a 5-year survival rate of 28% (14). Clinical and laboratory findings,
including negative molecular testing for the
JAK2V617F mutation, facilitated in the exclusion
of other common MPNs, such as polycythemia vera (PV), essential
thrombocythemia (ET) and primary myelofibrosis. However, this
mutation has been detected in 13 cases of CNL to date, indicating
that the JAK2V617F mutation is a rare event in
patients with atypical MPN (15).
Treatment approaches for individuals with this disorder are
heterogeneous.
The case presented in the current study was
characterized by marked and sustained neutrophilia, an increased
NAP score, absence of the Ph chromosome, a lack of
BCR/ABL1 fusion transcripts, as detected by RT-qPCR,
and no underlying disorders causing leukemoid reaction. Carcinoma
and infectious disease were eliminated during differential
diagnosis, no monoclonal gammopathy was identified, as in multiple
myeloma, and no complications, such as PV, ET or myelodysplastic
syndrome (MDS), were observed. In the present case,
hepatosplenomegaly was not detected in the patient upon initial
admission, however, other laboratory findings satisfied the WHO
diagnostic criteria for CNL. Due to progression of the disease, the
patient exhibited bleeding gums, splenomegaly and lymphadenopathy 1
year later. The following mean peripheral blood cell counts were
obtained during the patient's clinical course: A leukocyte count of
28.36×109/l (range, 7.67–51.28×109/l),
including 88.5% mature neutrophils (range, 78.8–98.5%), and a
platelet count of 33.3×109/l (range,
23–94×109/l). Thrombocytopenia was observed in the
patient upon presentation and followed a progressive course,
exhibiting no response to treatment with hydroxyurea and IFN-α.
Notably, the present study identified EVI-1 expression bone marrow
samples from the patient by performing qualitative and quantitative
screening tests. EVI-1 is an oncogene that confers poor prognosis
in human hematological malignancies, including acute myeloid
leukemia, CML and MDS (8). It was
initially identified as the integration site of the ecotropic
retrovirus that causes myeloid leukemia in murine model systems
(16). The significant function of
EVI-1 in hematopoietic stem cell regulation indicates that EVI-1
may participate in the generation of leukemic stem cells. These
leukemic stem cells are a potential cause of therapeutic resistance
in patients with leukemia (8).
Furthermore, increased expression of EVI-1 may be important in
development of human leukemias, particularly in the progression
from the chronic to blastic crisis phases of CML, and even in cases
without chromosome 3q26 abnormalities (17). In patients for whom the cause of
neutrophilia is not easily discernible, the incorporation of
CSF3R mutation testing may be a useful point-of-care
diagnostic tool to evaluate the presence of a clonal myeloid
disorder, as well as providing the potential for
genetically-informed treatment. Maxson et al (6) detected a CSF3R mutational
frequency of 89% in CNL and 44% in aCML, the most common of which
is the p.T618I membrane proximal mutation. The present patient's
results in the demonstrated a p.T618I membrane proximal mutation of
CSF3R in the CD15+ and CD34+ cell
fractions. Determining the presence and type of CSF3R
mutation is valuable, as they may be useful in the differential
diagnosis of leukocytosis and act as indicators of sensitivity to
different kinase inhibitors. Thus, categorizing the type of
CSF3R mutation in a patient with CNL/aCML promotes
individualized therapy. Pardanani et al (10) recently performed SETBP1
mutation screening in CNL/aCML cases and closely associated MNP
cases. Overall, SETBP1 mutational frequencies in WHO-defined
CNL, aCML, chronic myelomonocytic leukemia and PMF were 33, 0, 7
and 3%, respectively. By contrast, Piazza et al (9) recently identified SETBP1
mutations in 25% of aCML patients. Notably, Pardanani et al
(10) identified no significant
correlation between the presence of CSF3R mutations and age,
gender or leukocyte count. In addition, it was identified that the
presence or absence of CSF3R mutations did not affect
survival, whereas there was a trend for shortened survival among
SETBP1-mutated patients. Accordingly, the overexpression of
EVI-1 and the presence of a SETBP1 mutation identified in
the current patient are indicators of poor prognosis. Unusually,
genetic analysis of the current patient indicated concurrent
mutations of CSF3R and SETBP1. A previous study
identified that these two mutations are not mutually exclusive in
CNL/aCML. Follow-up sequencing of an expanded cohort of 29 patients
with CNL/aCML revealed that 21% exhibited CSF3R and
SETBP1 mutations, including 31% with CSF3R mutations
only and 7% with SETBP1 mutations only (7). These concurrent mutations indicate a
requirement for different therapeutic approaches tailored to the
molecular profile of individual patients (6). In the present study, there was
insufficient time to commence treatment with JAK2 inhibitors, such
as ruxolitinib, prior to the patient succumbing to the disease.
However, Lasho et al (18)
recently described a double-mutated CNL patient (CSF3R and
SETBP1) who was refractory to treatment with ruxolitinib and
hydroxyurea.
In conclusion, the present study reports the case of
a 67-year-old man who presented with sustained neutrophilia,
persistent thrombocytopenia, absent BCR-ABL1 transcripts and
a JAK2V617F mutation, in addition to
overexpression of EVI-1 and a novel, concurrent mutation of
CSF3R and SETBP1. The patient was ultimately
diagnosed with CNL. The current study demonstrates the difficulty
in establishing a correlation between EVI-1 overexpression and the
pathogenesis of CNL; therefore, additional investigations are
required to elucidate the mechanism by which EVI-1 is associated
with the pathogenesis of CNL.
Acknowledgements
This study was supported by the National Natural
Sciences Foundation of China (grant no. 81400092), the National
Natural Sciences Foundation of Tianjin (grant no. 13JCYBJC23400)
and the Science and Technique Foundation of Tianjin (grant no.
13KG106)
References
|
1
|
Bain BJ, Brunning BR, Vardiman JW and
Thiele J: Chronic neutrophilic leukaemiaWHO Classification of
Tumors of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon,
France: pp. 38–39. 2008
|
|
2
|
Böhm J and Schaefer HE: Chronic
neutrophilic leukaemia: 14 new cases of an uncommon
myeloproliferative disease. J Clin Pathol. 55:862–864. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elliott MA: Chronic neutrophilic leukemia
and chronic myelomonocytic leukemia: WHO defined. Best Pract Res
Clin Haematol. 19:571–593. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beekman R and Touw IP: G-CSF and its
receptor in myeloid malignancy. Blood. 115:5131–5136. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dong F, van Buitenen C, Pouwels K, et al:
Distinct cytoplasmic regions of the human granulocyte
colony-stimulating factor receptor involved in induction of
proliferation and maturation. Mol Cell Biol. 13:7774–7781.
1993.PubMed/NCBI
|
|
6
|
Maxson JE, Gotlib J, Pollyea DA,
Fleischman AG, Agarwal A, Eide CA, et al: Oncogenic CSF3R mutations
in chronic neutrophilic leukemia and atypical CML. N Engl J Med.
368:1781–1790. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gotlib J, Maxson JE, George TI and Tyner
JW: The new genetics of chronic neutrophilic leukemia and atypical
CML: Implications for diagnosis and treatment. Blood.
122:1707–1711. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goyama S and Kurokawa M: Pathogenetic
significance of ecotropic viral integration site-1 in hematological
malignancies. Cancer Sci. 100:990–995. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piazza R, Valletta S, Winkelmann N,
Redaelli S, Spinelli R, Pirola A, et al: Recurrent SETBP1 mutations
in atypical chronic myeloid leukemia. Nat Genet. 45:18–24. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pardanani A, Lasho TL, Laborde RR, Elliott
M, Hanson CA, Knudson RA, et al: CSF3R T618I is a highly prevalent
and specific mutation in chronic neutrophilic leukemia. Leukemia.
27:1870–1873. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vardiman J and Hyjek E: World Health
Organization classification, evaluation, and genetics of the
myeloproliferative neoplasm variants. Hematology Am Soc Hematol
Educ Program. 1:250–256. 2011. View Article : Google Scholar
|
|
12
|
Tuohy E: A case of splenomegaly with
polymorphonuclear neutrophil hyperleukocytosios. Am J Med Sci.
160:18–24. 1920. View Article : Google Scholar
|
|
13
|
Böhm J, Kock S, Schaefer HE and Fisch P:
Evidence of clonality in chronic neutrophilic leukaemia. J Clin
Pathol. 56:292–295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reilly JT: Chronic neutrophilic leukaemia:
A distinct clinical entity? Br J Haematol. 116:10–18. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Elliott MA and Tefferi A: The molecular
genetics of chronic neutrophilic leukaemia: Defining a new era in
diagnosis and therapy. Curr Opin Hematol. 21:148–154. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morishita K, Parker DS, Mucenski ML,
Jenkins NA, Copeland NG and Ihle JN: Retroviral activation of a
novel gene encoding a zinc finger protein in IL-3-dependent myeloid
leukemia cell lines. Cell. 54:831–840. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ogawa S, Kurokawa M, Tanaka T, Tanaka K,
Hangaishi A, Mitani K, et al: Increased EVI-1 expression is
frequently observed in blastic crisis of chronic myelocytic
leukemia. Leukemia. 10:788–794. 1996.PubMed/NCBI
|
|
18
|
Lasho TL, Mims A, Elliott MA, Finke C,
Pardanani A and Tefferi A: Chronic neutrophilic leukemia with
concurrent CSF3R and SETBP1 mutations: Single colony clonality
studies, in vitro sensitivity to JAK inhibitors and lack of
treatment response to ruxolitinib. Leukemia. 28:1363–1365. 2014.
View Article : Google Scholar : PubMed/NCBI
|