Introduction
Pineal region tumors, which account for <1% of
intracranial tumors (1), encompass a
heterogeneous group of neoplasms that may be divided into four main
categories: Germ cell tumors, glial cell tumors, pineal parenchymal
tumors (PPTs) and other miscellaneous tumors and cysts. PPTs
originate from cells in the pineal gland called pinealocytes, and
represent only 0.3% of all primary tumors of the central nervous
system (2). The current World Health
Organization classification of PPTs includes well-differentiated
pineocytoma (PC), PPT of intermediate differentiation and poorly
differentiated pineoblastoma (PB) (3).
PB is more common in children than in adults. It has
been reported that the peak incidence of PB occurs in the first 4
years of life, with tendency to arise in the first and second
decades (4). In addition, adult cases
of PB account for <10% of published cases (5). Due to the rarity of PB, relevant data is
limited, particularly regarding PB in adults. Thus, the biology,
standard management and prognosis of PB are not well understood at
present.
The current report describes a case of PB occurring
in a 46-year-old male who presented with obstructive hydrocephalus
due to a large pineal region mass. This case highlights a minimally
invasive strategy to treat a rare pineal region tumor with
significant involvement of critical structures in an adult. Written
informed consent was obtained from the patient.
Case report
A 46-year old male was admitted to West China
Hospital of West China Medical School (Sichuan University, Chengdu,
China) in June 2011 with a 3-month history of mild headache,
dizziness and impaired vision. No abnormalities were observed upon
physical examination and laboratory tests. An ophthalmological exam
revealed no evidence of papilledema.
Computed tomography (CT) of the brain was performed,
and a solid mass measuring 5 cm in diameter was observed in the
pineal region with notable obstructive hydrocephalus. Magnetic
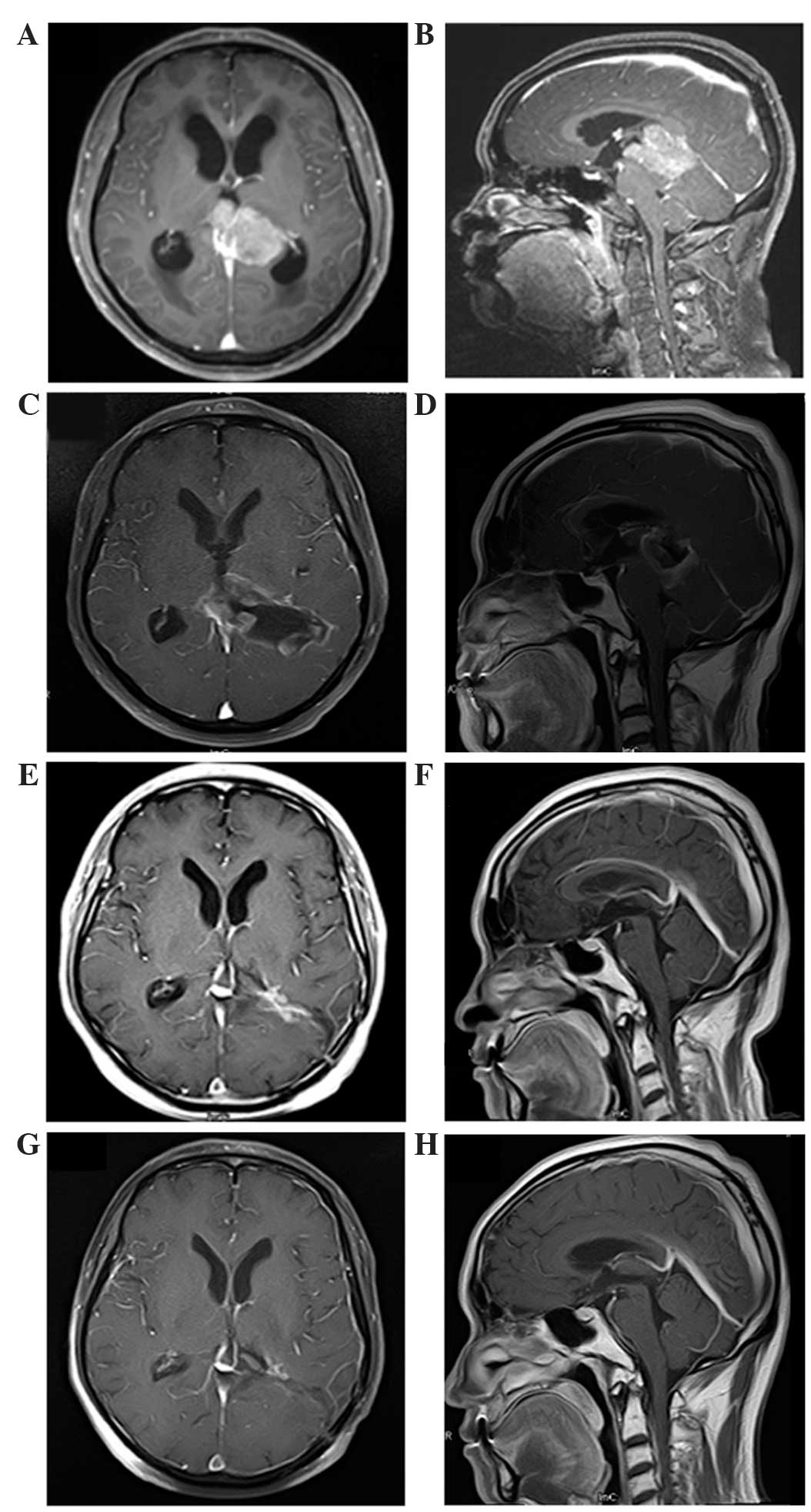
resonance imaging (MRI) of the brain was also performed (Fig. 1), revealing a large tumor measuring
4.1×5.1×3.9 cm in the pineal region. The mass was hypointense on
T1-weighted imaging and hyperintense on T2-weighted imaging, and
was heterogeneously enhanced following gadolinium administration.
The tumor was close to the midbrain and thalamus, and protruded
forward into the posterior part of the third ventricle and backward
into the cistern of the great cerebral vein. Both internal cerebral
veins and the great cerebral vein were enclosed within the tumor.
The mass was compressing the triangular region of the left lateral
ventricle from its lower and medial side, and the mesencephalic
aqueduct from its posterior side, and caused obstructive
hydrocephalus in the supratentorial ventricles (Fig. 1A and B). MRI of the spine revealed no
lesions. The differential diagnosis based on MRI included
germinoma, glial tumor and ependymoma. Tests for the tumor markers
β-human chorionic gonadotropin, α-fetoprotein and carcinoembryonic
antigen in the serum and cerebrospinal fluid (CSF) were negative.
CSF cytology was also negative.
Due to the location of the lesion and significant
involvement of critical structures, gross total resection (GTR) was
not adopted and partial removal of tumor was performed. Neural
guiding technology was used to locate the tumor precisely. In
addition, both internal cerebral veins and the great cerebral vein
were carefully identified and protected to a ensure minimally
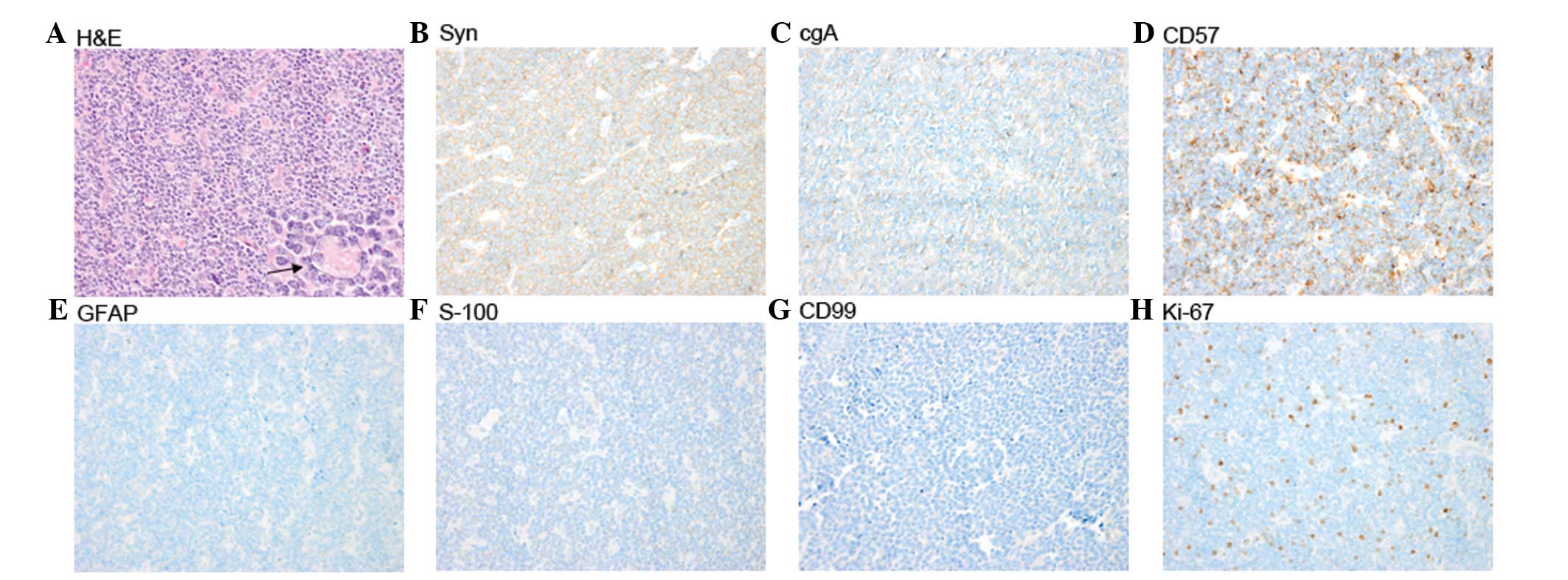
invasive procedure. Histological examination demonstrated a highly
cellular tumor composed of small, round cells that were darkly
stained, with hyperchromatic oval nuclei and scanty cytoplasm, and
which were partially arranged in Homer-Wright rosettes (Fig. 2A). Mitosis was also observed. The
majority of the tumor cells demonstrated immunoreactivity for
synaptophysin (Fig. 2B), chromogranin
A (Fig. 2C) and CD57 (Fig. 2D), and negativity for glial fibrillary
acidic protein (Fig. 2E), S-100
(Fig. 2F) and CD99 (Fig. 2G). The Ki-67 proliferation index was
~15% (Fig. 2H). Based on these
findings, a pathological diagnosis of PB was determined.
Immediate postoperative MRI revealed a residual mass
with gadolinium enhancement in the pineal region (Fig. 1C and D). The patient underwent
subsequent external beam radiation therapy for prophylactic
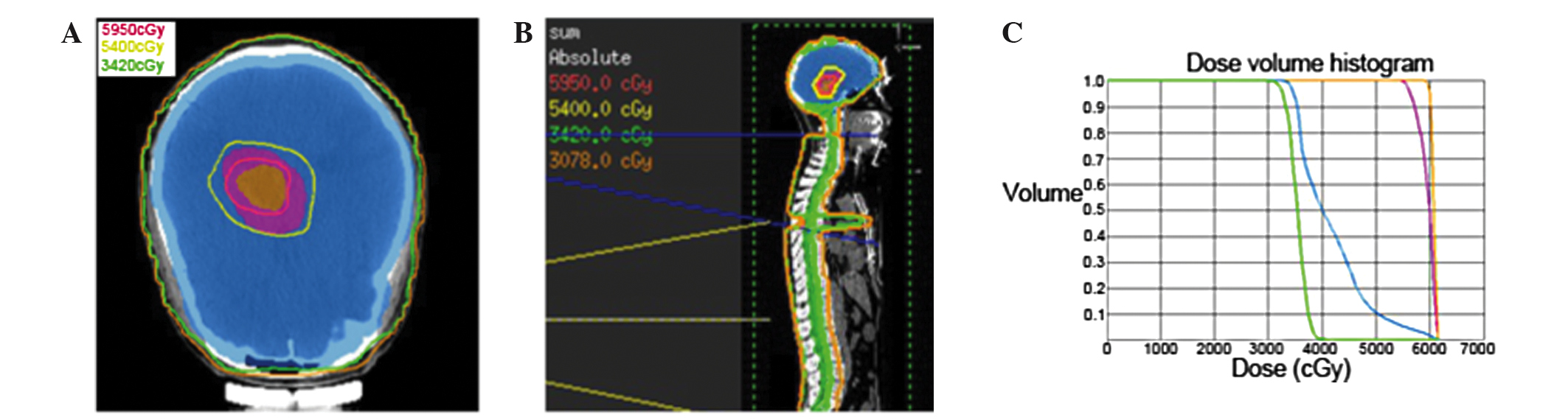
craniospinal irradiation (CSI) to 34.2 Gy, followed by a local
‘boost’ to the tumor site for a total of 59.5 Gy (Fig. 3A–C). The patient was treated with 6 MV
photons in a prone position in two phases. In phase 1, prophylactic
CSI was administered to a dose of 34.2 Gy in 19 fractions (1.8
Gy/fraction) over 23 days using a conformal technique. In phase 2,
a local ‘boost’ was administered to the residual gross tumor volume
(GTV) at a dose of 25.3 Gy in 11 fractions (2.3 Gy/fraction) and to
the clinical target volume (GTV plus a 1 cm margin) at a dose of
19.8 Gy in 11 fractions (1.8 Gy/fraction) over 15 days by
intensity-modulated radiotherapy. During radiotherapy, treatments
were tolerated extremely well by the patient, and no significant
side effects were experienced.
At 2 months post radiation, MRI demonstrated that
postoperative radiotherapy had resulted in complete regression of
the tumor (Fig. 1E and F). At the
36-month follow-up after radiation, the patient exhibited no signs
of tumor recurrence or neurological deficits (Fig. 1G and H).
Discussion
Adult PB is rare central nervous system tumor,
categorized as a supratentorial primitive neuroectodermal tumor
(PNET) localized to the pineal gland, with a propensity to
disseminate along the neuraxis and relapse (6). Despite certain similarities, patients
with PB have different structural and immunohistochemical
characteristics and such cases have poorer outcomes compared with
those of infratentorial PNETs (e.g., medulloblastomas). Cases of PB
in adults also have a poorer prognosis than those in children
(7). Little information has been
established with regard to the clinical features and outcomes of
adult patients with PB.
There is no consensus on the definitive CT and MRI
observations among the published cases of PB. On CT, PBs tend to be
large, lobulated and heterogeneously enhanced with infrequent
calcifications (8). In addition, PB
often presents with a greater degree of hydrocephalus compared with
PC. The tumors are typically solid and occasionally cystic,
although this is more frequently observed in PC (9). On MRI, PBs are usually characterized by
hypointensity or hypo to isointensity on T1-weighted images,
isointensity or iso to hyperintensity on T2-weighted images, and
heterogeneous enhancement in the pineal region (10). The current patient presented with a
number of these radiographic characteristics.
Histologically, PB is very similar to other PNETs
and has been described as ‘supratentorial PNET’ (6). PB is a highly cellular tumor composed of
small, round, poorly differentiated cells in patternless sheets or
aggregates. The cells contain hyperchromatic round or oval nuclei
and scanty cytoplasm and are usually arranged in Homer-Wright
rosettes, widely considered to represent abortive attempts at
neuroblastic differentiation (6).
Mitosis is frequently observed along with rosettes and areas of
necrosis (11). In addition, the
tumor cells typically demonstrate immunoreactivity for neuronal
markers, such as neurofilament, synaptophysin, chromogranin A,
glial fibrillary acidic protein and S-100 protein. In the current
report, the majority of the tumor cells demonstrated
immunoreactivity for synaptophysin and chromogranin A. The
pathological diagnosis of PB primarily depends upon the location
and morphology of the lesion. PB must be differentiated from other
tumor types located in the pineal region, including pineocytomas,
germ cell tumors and glial tumors.
The appropriate treatment strategies for PB have not
been determined as the incidence rate of PB is extremely low,
particularly in adults, and only a few described cases with limited
clinical follow-up and outcome studies are available (12–14).
Previous studies have reported that GTR may play a vital role in
the treatment of PB (15,16). However, despite significant
improvements in surgical techniques and perioperative care, a high
risk is associated with surgical intervention in the pineal area
owing to its proximity to critical structures. It has been reported
that the surgical mortality rate is 4–7% whilst the permanent
morbidity rate may be up to 10% (17). In the present case, the tumor was not
amenable to GTR due to its proximity to the midbrain and thalamus,
and because both internal cerebral veins and the great cerebral
vein were enclosed within the tumor. Considering quality of life, a
minimally invasive strategy of partial resection followed by
radiotherapy was adopted, which contributed to a favorable response
with complete tumor regression and an excellent neurological
outcome.
The function of postoperative therapy remains
undefined in PB. A few described cases reported that radiotherapy
aided in controlling the tumor and improving survival in patients
with PB (18,19). However, these benefits were mostly not
statistically assessed due to small sample size and the lack of
uniformity in radiotherapeutic strategies and doses. Lee et
al (16) examined treatment
factors that influenced survival in 34 adult patients who presented
with PB between 1969 and 1998, and found that the median survival
for patients who received ≥40 Gy of cranial irradiation was three
times that of patients receiving lower doses (29.8 vs. 8.1 months).
However, no prospective studies have confirmed the effect of
radiotherapy or optimal radiotherapeutic doses in PB to date.
The utilization of chemotherapy remains
controversial in PB. Hinkes et al (19) demonstrated partial responses to
chemotherapy in their series of six patients with PB. However, Lee
et al (16) reported that
chemotherapy did not confer any survival advantage among a series
of 34 adult PB patients of whom 10 underwent chemotherapy (16).
In the present case, the tumor volume was great and
its location was close to critical structures; therefore, partial
resection followed by radiotherapy was employed in order to avoid
nervous injury. Considering the tendency of PB to metastasize
widely throughout the CSF pathway, prophylactic CSI was
administered to a dose of 34.2 Gy. In addition, as PB is less
sensitive to radiotherapy than germinoma and medulloblastoma
(20,21), and the residual tumor volume following
surgery was large, a local ‘boost’ to the tumor site of 25.3 Gy in
2.3-Gy fractions was administered. The treatment resulted in
complete tumor regression without neurological deficits.
In conclusion, PB is rare in adults and, although
the appropriate treatment strategy for PB has yet to be determined,
the current case successfully demonstrates that aggressive surgery
must be avoided in patients whose tumors show significant
involvement of critical structures, taking into account surgical
complications, and that function-preserving resection followed by
postoperative radiotherapy may be the optimal treatment strategy.
However, prospective studies including a larger number of patient
groups are required to demonstrate the efficacy of chemo- and
radiotherapy and to determine the optimal standard treatment
strategy for PB in adults.
Acknowledgements
This study was supported by grants from Wu Jieping
Medical Foundation (no. 320.6750.13317), the program of Health
Department in Sichuan Province (no. 130087) and the program of
Science and Technology Development in Sichuan Province (no.
2014HH0063).
References
|
1
|
Al-Hussaini M, Sultan I, Abuirmileh N,
Jaradat I and Qaddoumi I: Pineal gland tumors: experience from the
SEER database. J Neurooncol. 94:351–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cuccia V, Rodríguez F, Palma F and Zuccaro
G: Pinealoblastomas in children. Childs Nerv Syst. 22:577–585.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nozza P, Casciana ML, Rossi A, Cama A,
Milanaccio C, Raso A, Ravegnani M, Morreale G and Garrè ML:
Post-chemotherapy maturation of a pineoblastoma. Acta Neuropathol.
119:651–653. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hart MN and Earle KM: Primitive
neuroectodermal tumors of the brain in children. Cancer.
32:890–897. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herrick MK and Rubinstein LJ: The
cytological differentiating potential of pineal parenchymal
neoplasms (true pinealomas). A clinicopathological study of 28
tumours. Brain. 102:289–320. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jakacki RI, Zeltzer PM, Boyett JM,
Albright AL, Allen JC, Geyer JR, Rorke LB, Stanley P, Stevens KR,
Wisoff J, et al: Survival and prognostic factors following
radiation and/or chemotherapy for primitive neuroectodermal tumors
of the pineal region in infants and children: A report of the
childrens cancer group. J Clin Oncol. 13:1377–1383. 1995.PubMed/NCBI
|
|
8
|
Chiechi MV, Smirniotopoulos JG and Mena H:
Pineal parenchymal tumors: CT and MR features. J Comput Assist
Tomogr. 19:509–517. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sugiyama K, Arita K, Okamura T, Yamasaki
F, Kajiwara Y, Ueda H and Kurisu K: Detection of a pineoblastoma
with large central cyst in a young child. Childs Nerv Syst.
18:157–160. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fujita A, Asada M, Saitoh M, Nakamura H,
Kamikawa S, Kokunai T and Tamaki N: Pineoblastoma showing unusual
ventricular extension in a young adult-case report. Neurol Med Chir
(Tokyo). 39:612–616. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schild SE, Scheithauer BW, Schomberg PJ,
Hook CC, Kelly PJ, Frick L, Robinow JS and Buskirk SJ: Pineal
parenchymal tumors. Clinical, pathologic and therapeutic aspects.
Cancer. 72:870–880. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
DeBoer R, Batjer H, Marymont M, Goldman S,
Walker M, GottardiLittell N and Raizer J: Response of an adult
patient with pineoblastoma to vorinostat and retinoic acid. J
Neurooncol. 95:289–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lutterbach J, Fauchon F, Schild SE, Chang
SM, Pagenstecher A, Volk B, Ostertag C, Momm F and Jouvet A:
Malignant pineal parenchymal tumors in adult patients: patterns of
care and prognostic factors. Neurosurgery. 51:44–56. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gururangan S, McLaughlin C, Quinn J, Rich
J, Reardon D, Halperin EC, Herndon J II, Fuchs H, George T,
Provenzale J, et al: High-dose chemotherapy with autologous
stem-cell rescue in children and adults with newly diagnosed
pineoblastomas. J Clin Oncol. 21:2187–2191. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tate M, Sughrue ME, Rutkowski MJ, Kane AJ,
Aranda D, McClinton L, McClinton L, Barani IJ and Parsa AT: The
long-term postsurgical prognosis of patients with pineoblastoma.
Cancer. 118:173–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee JY, Wakabayashi T and Yoshida J:
Management and survival of pineoblastoma: an analysis of 34 adults
from the brain tumor registry of Japan. Neurol Med Chir (Tokyo).
45:132–141; discussion 141–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Report of Brain Tumor Registry of Japan
(1969–1993). Neurol Med Chir (Tokyo). 40 (Suppl):S1–S106. 2000.
|
|
18
|
Chang SM, LillisHearne PK, Larson DA, Wara
WM, Bollen AW and Prados MD: Pineoblastoma in adults. Neurosurgery.
37:383–390; discussion 390–391. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hinkes BG, von Hoff K, Deinlein F,
Warmuth-Metz M, Soerensen N, Timmermann B, Mittler U, Urban C, Bode
U, Pietsch T, et al: Childhood pineoblastoma: experiences from the
prospective multicenter trials HIT-SKK87, HIT-SKK92 and HIT91. J
Neurooncol. 81:217–223. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reddy AT, Janss AJ, Phillips PC, Weiss HL
and Packer RJ: Outcome for children with supratentorial primitive
neuroectodermal tumors treated with surgery, radiation, and
chemotherapy. Cancer. 88:2189–2193. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Claude L, FaureConter C, Frappaz D,
Mottolèse C and Carrie C: Radiation therapy in pediatric pineal
tumors. Neurochirurgie. 61:212–215. 2015. View Article : Google Scholar : PubMed/NCBI
|