Introduction
Neuroendocrine tumors (NETs) are rare tumors arising
from enterochromaffin (Kulchitsky) cells, which are part of the
amine precursor uptake and decarboxylation system. The tumors are
thus found in locations with a wide distribution of these cells,
such as in the gastrointestinal tract, lungs and bronchi (1).
The World Health Organization (WHO) classification
of NETs groups the tumors into those of epithelial origin, namely
typical (well-differentiated) carcinoid (TC) tumors, atypical
(moderately-differentiated) carcinoid (AC) tumors and small cell
(undifferentiated) neuroendocrine carcinoma (SmCC), and those of
neural origin, namely paraganglioma (2).
The clinical and pathological features of NETs are
characteristic of the organ of origin, however, these tumors can
share other attributes irrespective of their anatomical site
(3). Although NETs may have a similar
presentation to other head and neck tumors in the same locations,
their behavior and management are not clearly established and vary
according to their histological type (4). Functioning tumors, particularly
carcinoid NETs, can present with symptoms caused by hormone
secretion; for example, patients may present with carcinoid
syndrome, which is characterized by flushing, diarrhea and
abdominal pain (3,5). A diagnosis of carcinoid NET is suspected
when features of carcinoid syndrome are present and high urinary
5-hydroxyindoleacetic acid levels are identified, with histological
analyses used to confirm the diagnosis (3,5).
Therapeutic strategy selection depends on the site of origin of the
tumor and its extent. Treatment usually consists of surgery and/or
radiotherapy with or without chemotherapy (6). Rarely, NETs occur in the head and neck
region, predominantly in the larynx and the middle ear, however,
few cases have been described in the nasopharynx (2).
In the present study, three cases of head and neck
NETs in the three different aforementioned locations are described
along with their management and follow-up.
Case report
Data collection
All head and neck NET cases treated in Hotel Dieu de
France Hospital (Beirut, Lebanon), between January 2004 and January
2014, were retrospectively reviewed. Data regarding clinical
presentation, management, pathology results and follow-up were
retrieved from hospital charts. A total of three cases were found.
Written informed consent was obtained from all patients prior to
surgery and prior to writing this study. A review of the English
and French literature regarding NETs of the head and neck region
was performed. This review was based on a search of the US National
Library of Medicine (PubMed) between 1990 and 2014.
Case 1
A 64-year-old Caucasian male presented in June 2007
with hoarseness and dysphagia that had persisted for 2 years. The
patient had smoked 2 packs of cigarettes per day for 20 years until
quitting 20 years ago. The patient denied a history of alcohol
consumption. The patient also experienced moderate dyspnea,
particularly when in the left lateral decubitus position. A total
of 20 kg of weight loss had been noted over the last 2 years. At 1
month prior to the current visit, the patient developed right-sided
otlagia. A laryngoscopy revealed a mass in the right aryepiglottic
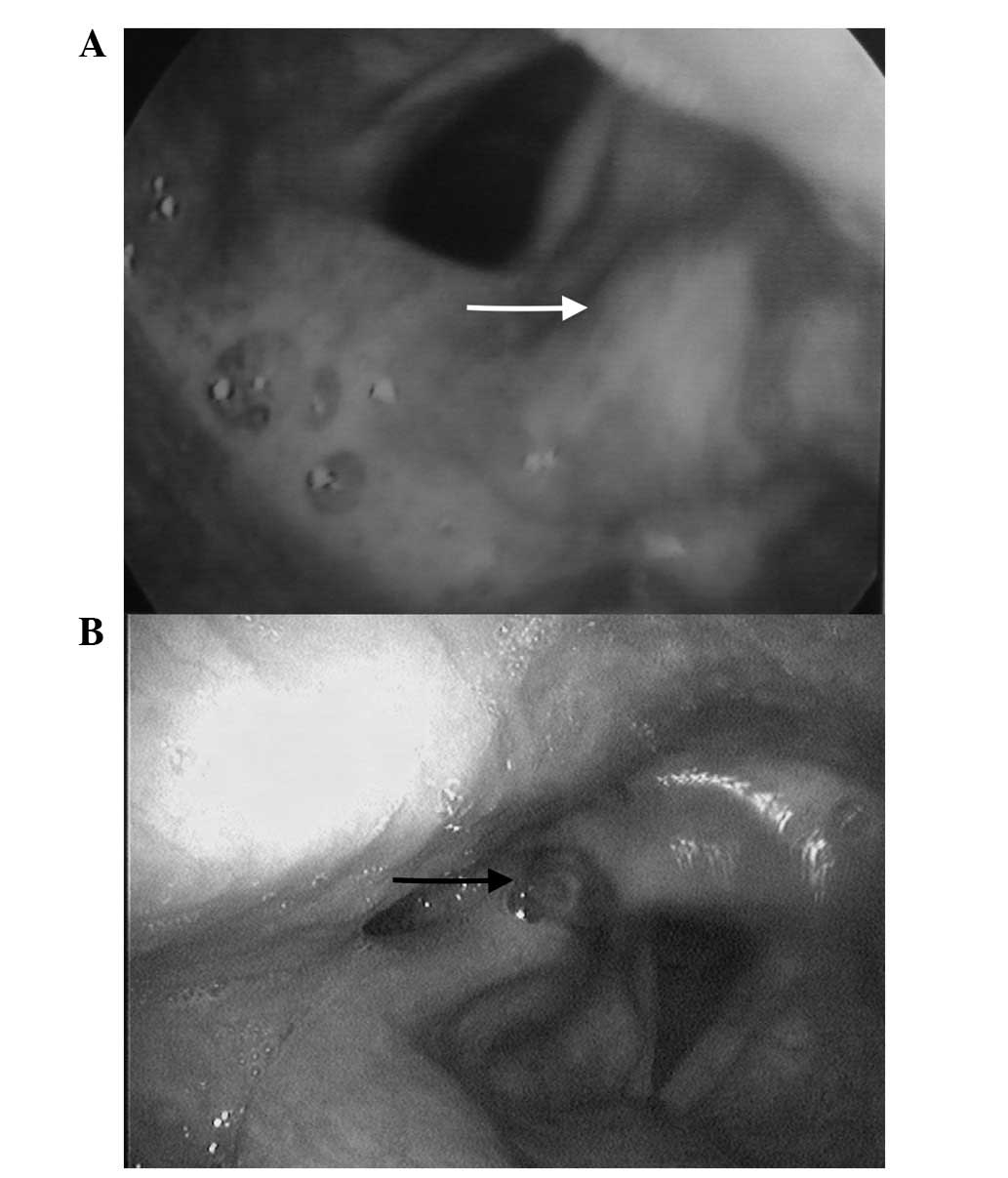
fold (Fig. 1A). An endoscopic
excisional biopsy of the lesion was performed. The tumor was
composed of tubular structures and compact cords. Moderate
anisonucleosis was present and the cells exhibited powder-like
chromatin. Few mitotic and apoptotic cells were noted. Stroma was
mildly abundant and endocrinoid in aspect. Periodic acid-Schiff
coloration showed no intracytoplasmic mucosecretions.
Immunohistochemistry was positive for chromogranin A, synaptophysin
and cytokeratin 7 (CK7). The final diagnosis was of an AC tumor of
the larynx in the right aryepiglottic fold.
Radiotherapy was suggested, but the patient refused
this therapeutic option. After 3 years, recurrence of the disease
was noted on the right arytenoid cartilage, which was treated by
endoscopic resection. The pathology report showed recurrence of the
AC tumor, with negative resection margins. The patient was then
followed up regularly. At the 5-month follow-up visit, a mass was
observed in the right arytenoid and aryepiglottic fold on direct
laryngoscopy (Fig. 1B). Endoscopic
excision of the lesion was performed using a CO2 laser.
The pathology report once again showed recurrence of the AC tumor,
with clean resection margins. The patient then underwent 28
sessions of adjuvant radiotherapy (56 Gy) for a total of 4 weeks
and is currently free of disease 9 months after this treatment.
Case 2
A 43-year-old Caucasian female presented in July
2006 with the sensation of ear fullness and mild hearing loss that
had persisted for 5 months, but with no otorrhea, vertigo or
tinnitus. A physical examination showed a white retro-tympanic mass
in the right ear and conductive hearing loss in the same ear,
confirmed by an audiogram. A computed tomography (CT) scan showed a
well-defined mass in the mesotympanum, with no erosion of the ear
ossicles. The patient subsequently underwent a radical
mastoidectomy with excision of the middle ear mass.
The tumor was composed of a tubular and trabecular
proliferation with no patent signs of cytological malignancy. The
immunohistochemistry was positive for chromogranin A and CK7. The
final diagnosis was of a TC tumor of the middle ear. No clinical
evidence of disease recurrence was found after 8 years.
Case 3
A 51-year-old Caucasian male presented in February
2013 with atypical vertigo that had first occurred 2 months
previously. The patient had smoked 2 packs of cigarettes per day
for 30 years and denied a history of alcohol consumption. No
additional complaints, other than an old history of chronic nasal
obstruction, were noted. Otoscopy, anterior rhinoscopy and an oral
cavity examination were normal, as was the neck palpation. Brain
magnetic resonance imaging was performed for the workup of the
vertigo, which showed no abnormal findings in the brain parenchyma,
but revealed the presence of a 2.2×2.8-cm mass located in the
nasopharynx and in contact with the posterior region of the nasal
septum. A CT scan was then performed and showed a 2.5-cm polyp-like
mass in the nasopharynx. The mass was of tissular density and
contained small calcifications, with no visible adenopathies on CT
scan. A subsequent endoscopic resection of the mass was
performed.
Histopathologically, the tumor was composed of an
endocrinoid proliferation containing sheets and nests separated by
a thin vascularized stroma. The tumor cells exhibited round nuclei
containing salt and pepper-like chromatin. Cytoplasm was abundant
and amphophilic. Mild nuclear pleomorphism was noted and punctuate
areas of necrosis were present. Mitotic activity was low with a
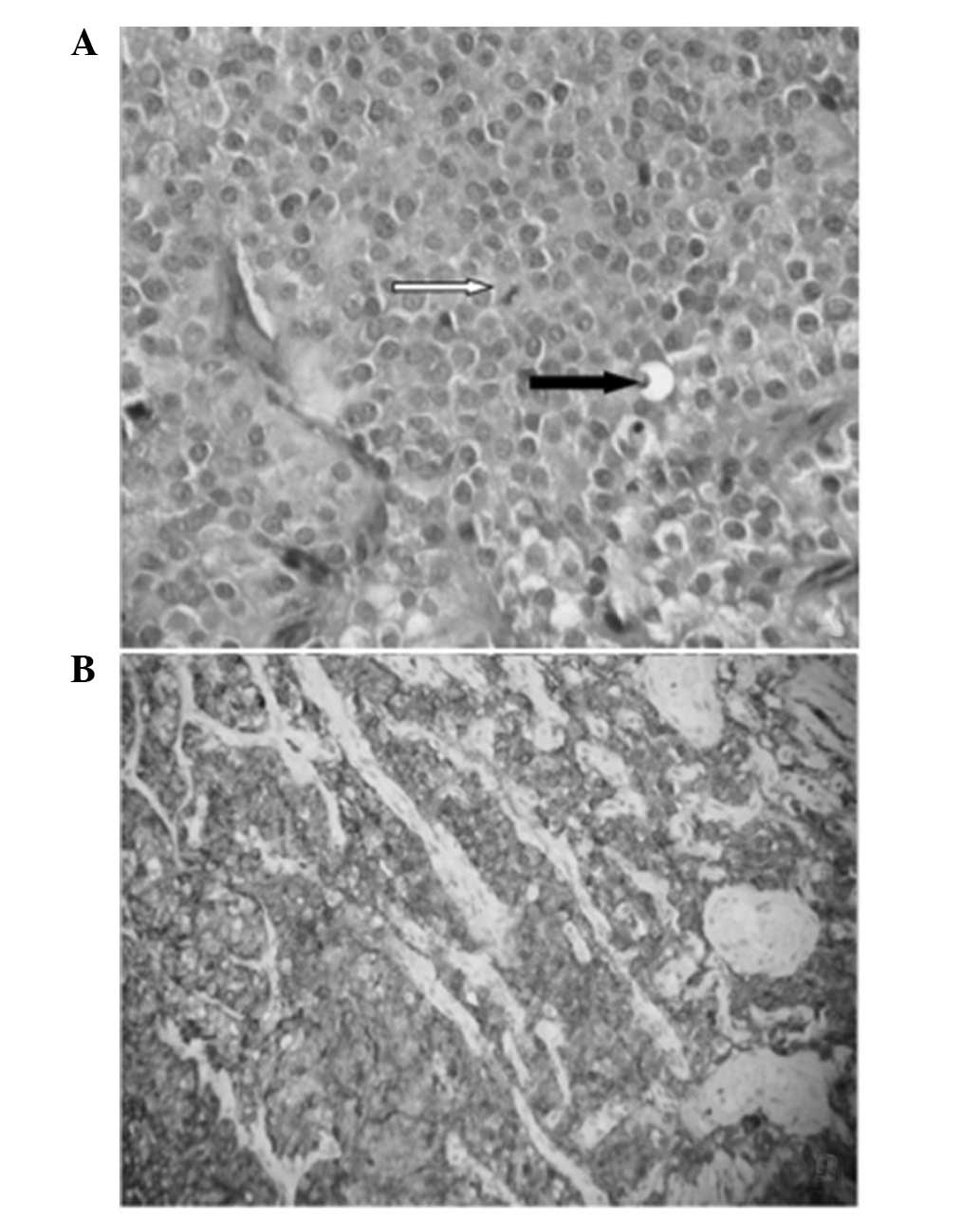
rate of 1/10 high-power fields (Fig.
2A). Immunohistochemistry was diffusely positive for
chromogranin A (Fig. 2B),
synaptophysin and pancytokeratin. There was no S100 staining of the
tumor cells, nor of the sustentacular cells. Calcitonin, thyroid
transcription factor-1 and caudal-type homeobox-2 staining was also
negative. The proliferation index was 3%. The final diagnosis was
of an AC tumor of the nasopharynx.
Positron emission tomography-CT scanning with
gallium-68-DOTA-NOC was performed 1 month post-operatively to
search for other localizations and revealed positive uptake in the
nasopharynx only. The urinary 5-hydroxyindoleacetic acid level was
9.1 mg/24 h (normal range, 2–6 mg/24 h). At 3 months post-surgery,
the patient was administered 28 sessions of adjuvant radiotherapy
(56 Gy) for a total of 4 weeks, and is currently free of disease at
14 months of follow-up.
Discussion
Neuroendocrine neoplasms are rare tumors that are
mainly found in the GI tract, pancreas and lungs (5).
Numerous proposals have been put forward with regard
to the classification and nomenclature of NETs, and a number of
these differ in their use of specific terminology and the criteria
for grading and staging (3). In order
to predict the patient outcome and therapy, the use of a single
system of nomenclature, grading and staging is required for NETs of
all anatomical sites, as there are a number of similarities among
NETs throughout the body. However, certain systems that have arisen
independently have become firmly established and are recognized by
organizations charged with standardizing terminology, such as the
World Health Organization (WHO). No data favors one system over
another (3). The basic data that
underlie the systems are similar, even if the criteria differ.
NETs are rarely found in the head and neck,
particularly the larynx, the middle ear and the nasopharynx. The
WHO classification of laryngeal and middle ear NETs is generally
consistent with that for pulmonary neuroendocrine carcinoma. The
tumors can be subdivided into TC, AC, SmCC, combined SmCC with
non-small cell carcinoma, and those with neurological origins,
termed paragangliomas, according to the 2005 WHO classification of
head and neck tumors (2,6–8). With
regard to large cell neuroendocrine carcinomas, while the WHO
classification does not distinguish them from AC, they have been
considered as a separate subtype by one previous study (6). However, NETs of the nasopharynx are not
included in any of the WHO classifications of nasopharyngeal
tumors.
Histological grading depends on the number of
mitoses, the presence of necrosis and the Ki-67 index (Table I) (2,3,6,7).
 | Table I.Diagnostic criteria of neuroendocrine
tumors in the head and neck region. |
Table I.
Diagnostic criteria of neuroendocrine
tumors in the head and neck region.
| Criteria | TC | AC | SmCC |
|---|
| Neuroendocrine
differentiation | Yes | Yes | Yes |
| Mitotic count | 0–1/10 HPFs | 2–10/10 HPFs | >10/10 HPFs |
| Ki-67 | <2% | 3–20% | >20% |
| Cytoplasm amount | Moderate | Moderate | Little/scanty |
| Nucleoli | Inconspicuous | Inconspicuous | Inconspicuous |
| Nuclear
polymorphism | Little | Moderate | Moderate |
| Necrosis | No | Focal punctate or
mild | Marked |
In the head and neck region, NETs are mostly found
in the larynx. To date, ~650 patients with neuroendocrine neoplasms
of the larynx have been reported in the literature (9). The most frequent type is the AC tumor,
followed by SmCC, paraganglioma and finally, the TC tumor (10).
NETs rarely occur in the middle ear, but this
location is considered second in frequency after the larynx when it
comes to the head and neck region. Approximately 50 cases (11,12) of
carcinoid tumor of the middle ear have been reported in the English
literature (11). However,
neuroendocrine cells have not been demonstrated specifically within
the middle ear, and the precise origin of carcinoid tumor of the
middle ear has not been clarified (11).
NETs can also rarely occur in the nasal cavity or
the nasopharynx. In the nasopharynx, cases reported in the
literature were either SmCC or TC, with no cases of AC previously
reported. To the best of our knowledge, case 3 of the present study
is the first described case of a nasopharyngeal AC tumor.
The mainstay therapy for all NETs of the larynx,
with the exception of SmCC, is surgical excision. Depending on the
site and extent of the primary tumor, a partial or total
laryngectomy may be performed (13).
In a recent systematic review and meta-analysis of all laryngeal
neuroendocrine carcinomas, van der Laan et al reviewed
treatment schemes according to tumor subtypes and resulting
survival rates. The study found that the more benign end of the
spectrum is represented by TC tumors, and that local excision alone
is curative. Radiotherapy does not induce a good response in
patients with AC tumors, which are therefore best managed by
radical surgical excision in combination with elective bilateral
neck dissection due to the high propensity for regional metastasis.
The most benefit for cases of SmCC or large cell neuroendocrine
carcinoma appears to be gained from a combination of radiotherapy
and chemotherapy, although patient survival remains poor (14).
Laryngeal paragangliomas are almost always benign
and should be treated accordingly. Partial laryngectomy is
preferable to radiation as a cure is usually achieved without loss
of laryngeal function (15). Laser
surgery is not widely used due to the vascular nature of these
tumors (13).
Carcinoid tumors of the middle ear are primarily
treated by surgical excision. There is no known established
surgical approach, perhaps owing to the limited incidence of the
condition, but complete removal by tympano-mastoidectomy or radical
mastoidectomy is the most commonly used technique (16). In the literature, middle ear carcinoid
tumors were successfully removed by tympanotmy alone in certain
studies, while in other cases, a subtotal petrosectomy was
performed (17). No sufficient data
exist regarding the role of chemotherapy or radiation therapy in
middle ear carcinoid tumors (17).
Radiation therapy was used as adjuvant therapy in certain studies,
but the number of cases was insufficient to draw a conclusion
concerning better local control and recurrence rates (18).
In view of the small number of cases, no clear
treatment policy has been established for neuroendocrine carcinomas
of the nasal cavity, paranasal sinuses and nasopharynx. In almost
all the cases reported in the English literature, the treatment
consisted of surgery (7,19–25). In
certain studies, conventional radiotherapy was used alone or in
combination with either surgery or chemotherapy, but the efficacy
of such regimens is yet to be proven (22). In a review by Furuta et al, it
was proposed that radiation therapy and chemotherapy should be
performed as post-operative adjuvant therapy when a complete
resection is difficult and when surgery is ineffective (24).
NETs are rarely diagnosed in the head and neck
region. The tumors are found mainly in the larynx, less frequently
in the middle ear and scarcely in the nasal cavity or nasopharynx.
While a well-defined classification exists for NETs of the larynx
and the middle ear, these tumors are not mentioned in the most
recent WHO classification of nasopharyngeal tumors. Clinicians
should be aware of this tumor in the nasopharynx and should
consider it in the differential diagnosis of tumors located there.
Following the recent meta-analysis by van der Laan et al
(14), treatment schemes for
laryngeal NETs have been well established considering the low level
of evidence on this subject. However further studies are required
to establish treatment protocols for the middle ear and
nasopharyngeal locations.
References
|
1
|
Bapat U, Mackinnon N and Spencer MG:
Carcinoid tumours of the larynx. Eur Arch Otorhinolaryngol.
262:194–197. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barnes L, Eveson J, Reichart P and
Sidransky DE: Neuroendocrine tumoursWorld Health Organization
Classification of Tumours: Pathology and Genetics of Head and Neck
Tumours. IARC Press; Lyon: pp. 135–139. 2005
|
|
3
|
Klimstra DS, Modlin IR, Coppola D, Lloyd
RV and Suster S: The pathologic classification of neuroendocrine
tumors: A review of nomenclature, grading and staging systems.
Pancreas. 39:707–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meacham R, Matrka L, Ozer E, Ozer HG,
Wakely P and Shah M: Neuroendocrine carcinoma of the head and neck:
A 20-year case series. Ear Nose Throat J. 91:E20–E24.
2012.PubMed/NCBI
|
|
5
|
Kunz PL: Carcinoid and neuroendocrine
Tumors: Building on success. J Clin Oncol. 33:1855–1863. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kao H-L, Chang W-C, Li W-Y, Chia-Heng Li A
and Fen-Yau Li A: Head and neck large cell neuroendocrine carcinoma
should be separated from atypical carcinoid on the basis of
different clinical features, overall survival and pathogenesis. Am
J Surg Pathol. 36:185–192. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Weinreb I and Perez-Ordoñez B: Non-small
cell neuroendocrine carcinoma of the sinonasal tract and
nasopharynx. Report of 2 cases and review of the literature. Head
Neck Pathol. 1:21–26. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
LewisJS Jr, Ferlito A, Gnepp DR, Rinaldo
A, Devaney KO, Silver CE and Travis WD: International Head and Neck
Scientific Group: Terminology and classification of neuroendocrine
neoplasms of the larynx. Laryngoscope. 121:1187–1193. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ferlito A and Rinaldo A: The spectrum of
endocrinocarcinomas of the larynx. Oral Oncol. 41:878–883. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferlito A, Devaney KO and Rinaldo A:
Neuroendocrine neoplasms of the larynx: Advances in identification,
understanding and management. Oral Oncol. 42:770–788. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tabuchi K, Aoyagi Y, Uemaetomari I, Tobita
T, Wada T, Inadome Y, Noguchi M and Hara A: Carcinoid tumours of
the middle ear. J Otolaryngol Neck Surg. 38:E91–E94. 2009.
|
|
12
|
Bittencourt AG, Tsuji RK, Cabral F Junior,
Pereira LV, Fonseca AC, Alves V and Bento RF: Middle ear adenoma
with neuroendocrine differentiation: Relate of two cases and
literature review. Int Arch Otorhinolaryngol. 17:340–343.
2013.PubMed/NCBI
|
|
13
|
Ferlito A, Silver CE, Bradford CR and
Rinaldo A: Neuroendocrine neoplasms of the larynx: An overview.
Head Neck. 31:1634–1646. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van der Laan TP, Plaat BE, van der Laan BF
and Halmos GB: Clinical recommendations on the treatment of
neuroendocrine carcinoma of the larynx: A meta analysis of 436
reported cases. Head Neck. 37:707–715. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Myssiorek D, Rinaldo A, Barnes L and
Ferlito A: Laryngeal paraganglioma: An updated critical review.
Acta Otolaryngol. 124:995–999. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baig S, Patil N and Considine N: Carcinoid
tumour of the middle ear. J Coll Physicians Surg Pak. 22:604–606.
2012.PubMed/NCBI
|
|
17
|
Ramsey MJ, Nadol JB Jr..Pilch BZ and
McKenna MJ: Carcinoid tumor of the middle ear: Clinical features,
recurrences and metastases. Laryngoscope. 115:1660–1666. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krouse JH, Nadol JB and Goodman ML:
Carcinoid tumors of the middle ear. Ann Otol Rhinol Laryngol.
99:547–552. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Galm T and Turner N: Primary carcinoid
tumour of nasal septum. J Laryngol Otol. 123:789–792. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanoh N, Nishimura Y, Nakamura M, Mori M
and Uematsu K: Primary nasopharyngeal paraganglioma: A case report.
Auris Nasus Larynx. 18:307–314. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li AL, Wehrli B and Rotenberg BW:
Carcinoid tumour arising simultaneous to an inverted papilloma in
the nasal cavity. J Otolaryngol Head Neck Surg. 39:E78–E82.
2010.PubMed/NCBI
|
|
22
|
Vandist V, Deridder F, Waelput W, Parizel
PM, Van de Heyning P and Van Laer C: A neuroendocrine tumour of the
sphenoid sinus and nasopharynx: A case report. B-ENT. 6:147–151.
2010.PubMed/NCBI
|
|
23
|
Chu MW, Karakla DW, Silverberg M and Han
JK: Primary carcinoid tumor of the frontal sinus: A case report.
Ear Nose Throat J. 89:E13–E16. 2010.PubMed/NCBI
|
|
24
|
Furuta A, Kudo M, Kanai K, Ohki S and
Suzaki H: Typical carcinoid tumor arising in the nose and paranasal
sinuses-case report. Auris Nasus Larynx. 37:381–385. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee DH, Cho HH and Cho YB: Typical
carcinoid tumor of the nasal cavity. Auris Nasus Larynx.
34:537–539. 2007. View Article : Google Scholar : PubMed/NCBI
|