Introduction
Inflammatory myofibroblastic tumor (IMT) is a
relatively rare disease that predominantly occurs in the
peritoneum, retroperitoneum and lungs of young people; a previous
study of 38 cases reported a median age of 8.5 years (1). This disease is histologically
characterized by the proliferation of various spindle cells, which
are of mesenchymal cell origin, in myxoid or collagenous stroma
with prominent inflammatory infiltrates (2). IMT cells can potentially behave as
locally invasive and are rarely metastatic (in <5% of cases)
(3). In recent studies, a chromosomal
translocation involving 2p23 and subsequent anaplastic lymphoma
kinase (ALK) gene rearrangement was identified in ~50% of IMT cases
(3,4).
The ALK inhibitor, crizotinib, was demonstrated to be an effective
therapy for ALK rearrangement-positive IMT cases.
Complete surgical resection is typically conducted
for localized IMTs (5); however, no
standard therapeutic strategy has been established for inoperable
or metastatic IMTs. In the present study, an extremely rare case of
metastatic IMT that responded successfully to combination
chemotherapy of doxorubicin and ifosfamide was reported.
Case report
A 64-year-old male presented with abdominal
fullness, fever and severe fatigue in March 2014. These symptoms
had worsened within half a month. Following a visit to a primary
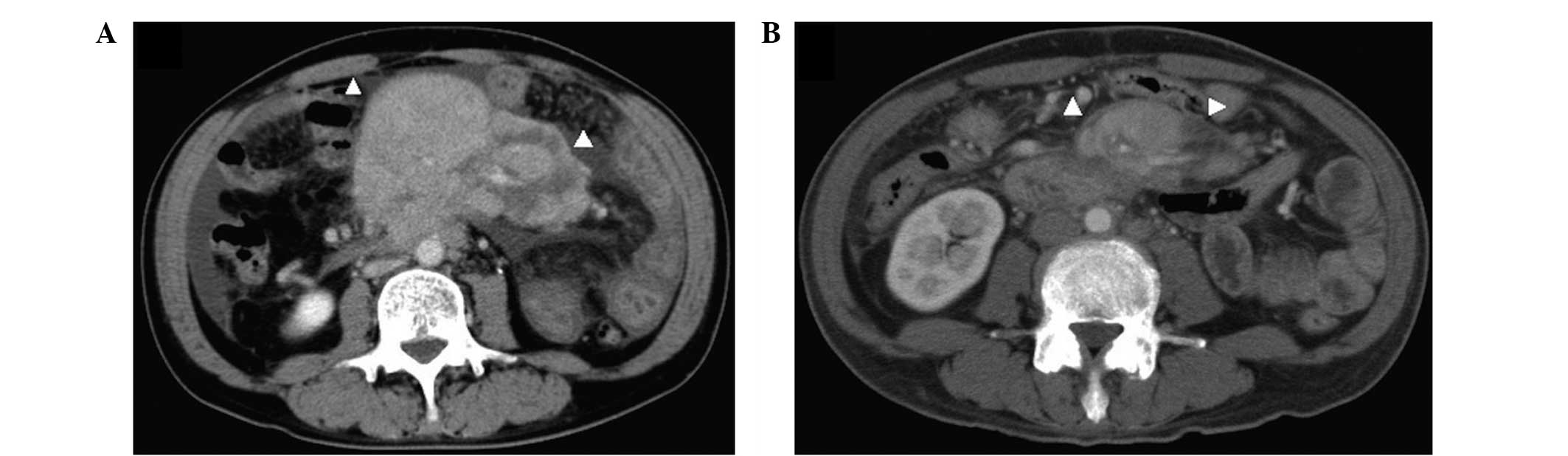
care doctor in March 2014, a computed tomography (CT) scan revealed
a moderate volume of ascites and a mass in the middle of the
abdomen (Fig. 1A). Initially,
lymphoma was suspected, and oral prednisolone (60 mg/day) was
administered for 3 days as a therapy with diagnostic intent;
however, no relief of the symptoms was observed. Exploratory
laparotomy indicated a protruded large tumor in the mesenterium and
2,500 ml of chylous ascites. Microscopic examination of the tumor
biopsy sample was unable to establish a diagnosis. In April 2014,
the patient was referred to the Department of Hematology and
Oncology at Kyushu University Hospital (Fukuoka, Japan) for further
evaluation. At the time of admission, the Eastern Cooperative
Oncology Group performance status (6)
had improved from grade 3 to grade 1 following removal of the
ascites, and the patient suffered from low back pain, fatigue and
fever. The laboratory results were as follows: C-reactive protein,
9.25 mg/dl (normal, ≤0.14 mg/dl); erythrocyte sedimentation, 105
mm/h (normal, 2–10 mm/h); platelet count, 45.6×104/µl [normal,
(158–348)×103/µl]; interleukin-6 (IL-6) level, 140 pg/ml (normal,
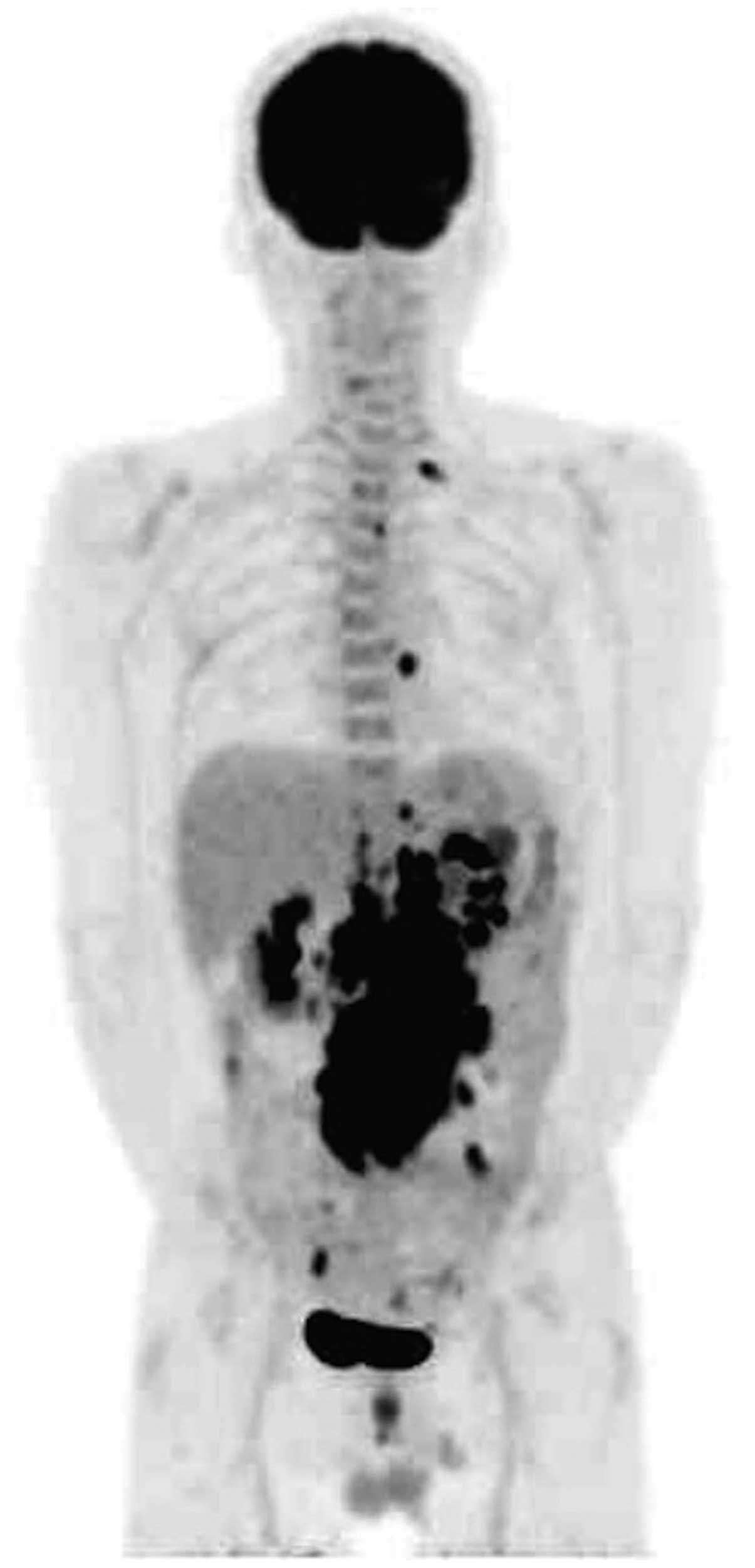
≤4.0 pg/ml). Fluorodeoxyglucose-positron emission tomography
(FDG-PET)/CT revealed hypermetabolic masses in the mesenteric
region [maximum standardized uptake value (SUVmax), 23.2], as well
as in the celiac artery and para-aortic region (SUVmax, 18.5), the
dorsal region of the thoracic descending aorta (SUVmax, 7.4) and
the left supraclavicular region (SUVmax, 5.8; Fig. 2). Magnetic resonance imaging (MRI)
examination indicated tumors with mixed high- and low-intensity
signals in a T2-weighted image (T2-WI).
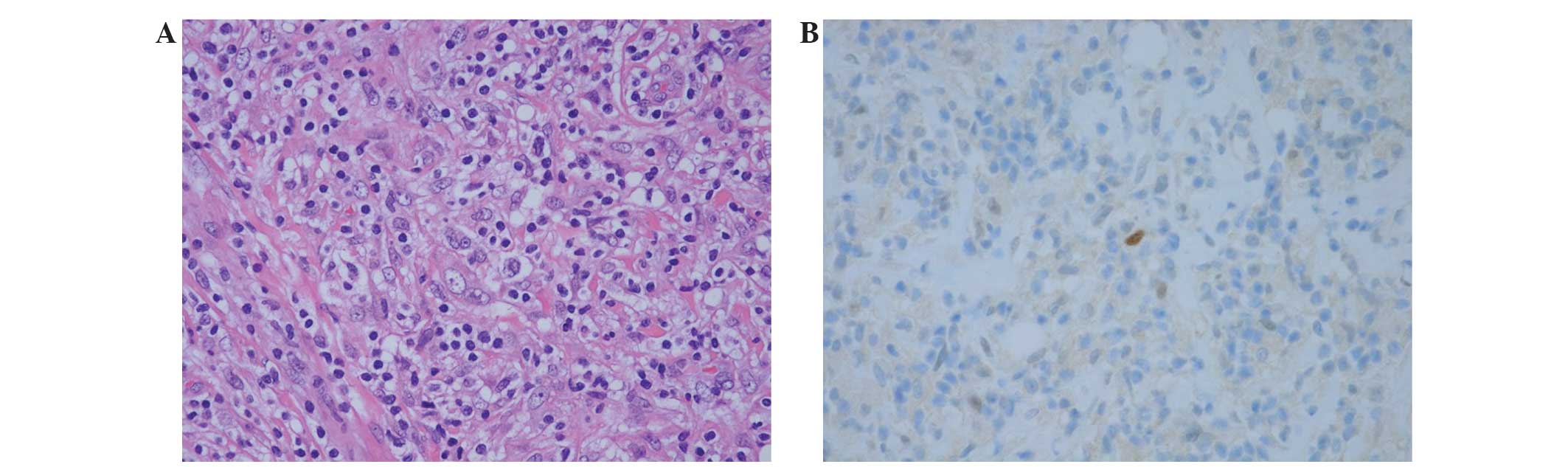
Intensive histological examination of the previously
obtained tumor sample demonstrated the proliferation of mildly
atypical cells that possessed enlarged, spindle to polygonal shaped
nuclei and vesicular chromatin arranged in a haphazard pattern.
These tumor cells were accompanied by an infiltration of
lymphocytes and plasmacytes (Fig.
3A). Immunohistochemical staining of the atypical tumor cells
was categorized as follows: Positive, >10% cells stained,
moderate-high intensity; negative, 0% cells stained; or focally
positive, not within the criteria for positive or negative. The
analyses revealed that the cells were positive for CD68, focally
and weakly positive for murine double minute 2 (MDM2; Fig. 3B) and focally positive for S-100
protein; by contrast, the cells were negative for α-smooth muscle
actin, cyclin-dependent kinase 4 (CDK4), ALK and CD35. The tumor
was also negative for Epstein-Barr virus-encoded small RNAs as
assessed by in situ hybridization. In addition, the plasmacytes
were negative for immunoglobulin G4 (IgG4). These histological
features suggested a type of inflammatory pseudotumor (IPT)-like
lesion; however, establishing a more accurate diagnosis was
difficult. The differential diagnosis included IMT, IPT-like
dedifferentiated liposarcoma (DLS).
At 7 days after admission, epigastrium and back
pain, a high fever and significant elevation of serum pancreatic
and biliary enzyme levels [lipase, 712 U/l (normal, 16–51 U/l);
total bilirubin, 3.5 mg/dl (normal, 0.4–1.5 mg/dl); alkaline
phosphatase, 1,553 U/l (normal, 106–322 U/l); γ-glutamyl
transpeptidase, 1,070 U/l (normal, 13–64 U/l)] were manifested. An
MRI scan revealed tumor invasion of the pancreas head causing
severe stenosis of the main pancreatic and choledochal duct.
Catheterization was then performed and the tumor exhibited rapid
growth and invaded to the adjacent tissue, which suggested a
malignant neoplasm.
Although a diagnosis could not be confirmed based on
the histological features alone, the patient was finally diagnosed
with an IMT based on the aforementioned clinical and histological
findings, including inflammatory symptoms and radiological images.
The patient received doxorubicin (15–25 mg/m2, days 1 and 2) and
ifosfamide (1.75–2 g/m2, days 1–5) every 3–4 weeks (AI therapy),
beginning in May 2014. A reduced dosage of chemotherapy was used
due to liver dysfunction and to avoid a fatal adverse event, such
as febrile neutropenia, in the subsequent cycle. Following the 4th
cycle of chemotherapy, a CT scan revealed a 50% reduction in the
baseline sum of the longest diameter of the target regions (from
12.5 cm to 6.2 cm), no size changes of the masses (all 6–7 mm in
the minor axis; these had been hypermetabolic on previous FDG-PET
in the celiac artery and para-aortic region, the dorsal region of
the thoracic descending aorta and the left supraclavicular region),
the disappearance of ascites and the absence of new lesions, which
was classified as partial response according to the Response
Evaluation Criteria in Solid Tumors (7) (Fig. 1B).
The patient experienced neutropenia (grade 4; according to Common
Terminology Criteria for Adverse Events version 4.0) (8), febrile neutropenia (grade 3) and sepsis
by Enterococcus cloacae (grade 4) during the 1st cycle, as well as
anorexia (grade 2) and fatigue (grade 1) for 5–6 days following the
initiation of each cycle of treatment. In addition, the patient did
not present any tumor-associated symptoms and the laboratory data,
including C-reactive protein level, erythrocyte sedimentation,
platelet count and IL-6 level, were within the normal limits. The
patient was planned to continue the chemotherapy up to a maximum of
7 cycles due to the dose-limiting cardiac toxicity of
doxorubicin.
Around the 5th cycle of AI therapy, radiological
examination revealed a moderate amount of ascites without any
change in the size of other lesions. From the 6th cycle, oral
prednisolone (30 mg/day) was initiated in combination with the
ongoing therapy, however, there was no change in the amount of
ascites. Following the 7th cycle of AI therapy, multiple new liver
metastatic lesions and hypermetabolic masses in the mediastinum,
axilla and intraabdominal region were revealed by FDG-PET. For
second-line chemotherapy, the patient was administered with a
combination therapy of gemcitabine and docetaxel, commencing in
November 2014, which was ineffective. The patient succumbed to
liver failure at around 7 months after the start of AI therapy.
The patient provided written informed permission for
the publication of this case.
Discussion
Inflammatory pseudotumors (IPTs) include a wide
variety of tumors from benign to malignant neoplasms, such as the
calcifying fibrous tumor, IgG4-associated inflammatory tumor,
IPT-like follicular dendritic cell sarcoma, IMT-like DLS and IMT.
Although it is occasionally difficult to histologically
differentiate between such tumors, the nature of these diseases is
diverse, particularly in terms of etiology (3). A chromosomal translocation involving
2p23 and subsequent ALK gene rearrangement was previously
identified in ~50% of IMT cases; however, older patients with IMT
tend to be negative for this translocation and gene rearrangement
(3,4).
While detection of ALK gene rearrangement in IPTs yields the
primary evidence for the diagnosis of IMT, a definitive diagnosis
is frequently difficult to achieve in cases of IMT without ALK gene
rearrangement (3). The present case
exhibited the following pathohistological features that were
characteristic of IPT: i) proliferation of mildly atypical spindle
cells; ii) low frequency of mitotic figures; and iii) prominent
infiltration of various inflammatory cells, including plasma cells
and lymphocytes. In addition, the patient suffered from severe
general fatigue and fever, and blood tests revealed a prominent
elevation of the inflammatory response; these features are
compatible with a diagnosis of IMT (1). CT and MRI scans demonstrated that the
tumors originated from the soft tissue of the abdomen and presented
a combination of high and low signal intensities on a T2-WI. The
region of low signal intensity on the T2-WI was enhanced in the
delayed phase of the CT scan, which indicates a fibrotic region,
and this finding is also consistent with IMT.
The greatest differential diagnosis may be DLS,
which rarely exhibits similar pathohistological characteristics
with IMT (9). DLS is often diagnosed
in older adults, as in the present case. Considering the
heterogeneity of the tumor, DLS cannot be definitively excluded
from the diagnosis due to the relatively small size of the biopsy
sample. A recent immunohistochemical study of DLS samples revealed
strong positive staining for MDM2 and CDK4 due to amplification of
chromosome 12q13–15 and the subsequent co-amplification of the MDM2
and CDK4 genes (10). MDM2 gene
amplification has also been detected in 27% of IMT cases (11). Therefore, the focally- and
weakly-positive staining of MDM2 and the negative staining of CDK4
in the present case could suggest a diagnosis of DLS; however, this
evidence could not confirm or exclude DLS. Considering both the
clinical and pathohistological findings, the current patient was
finally diagnosed with IMT.
The primary therapy for IMT is complete resection of
the tumors (5). However, this therapy
may not be suitable in the present case due to the large size of
the tumor surrounding large vessels, as well as due to the presence
of metastatic regions. Although the number of previous reports on
IMT is limited, a small number of cases have been reported
(5,12–16). A
study reporting a case with ALK gene rearrangement-positive IMT
demonstrated the failure of AI therapy followed by maintenance with
imatinib subsequent to surgery (12).
Another case of retroperitoneal IMT underwent systemic chemotherapy
with vincristine plus methotrexate and radiation, and no recurrence
was observed for 2 years (5). In
addition, certain studies on IMT cases in children reported the use
of various chemotherapeutic drugs or regimens, including
methotrexate, vinblastine, cisplatin, doxorubicin and ifosfamide,
which demonstrated variable responses and thus their efficacy is
unclear (13,17,18).
Non-steroidal anti-inflammatory drugs (NSAIDs) and corticosteroids
have also been studied as treatment options for IMT. Vascular
endothelial growth factor (VEGF) and cyclooxygenase (COX)2 are
highly expressed in the infiltrating inflammatory cells of IMT
(14). The COX2 inhibitor is
considered to suppress angiogenesis in the tumor tissue via
interference with the COX2/prostaglandin/VEGF axis. Two abdominal
IMT cases were reported to be responsive to NSAIDs (15). Systemic chemotherapy was administered
for the present patient due to the effects of the aggressive tumor
on the surrounding organs, including obstructive jaundice and
chylous ascites. The combination chemotherapy of doxorubicin and
ifosfamide that was employed in the current study was based on the
chemotherapy regimen for unresectable non-small round cell soft
tissue sarcomas (19). The 4th cycle
of the chemotherapy induced 50% reduction in the tumor diameter,
suggesting the effectiveness of the therapy. Celecoxib (400 mg per
day) was also administered from the initiation of chemotherapy
since the patient suffered from back pain, while dexamethasone (20
mg/day, days 1–6 of AI therapy) was used for prophylaxis of emesis.
There is a possibility that these additional agents may have also
affected the suppression of tumor growth.
The kinase activity of the ALK fusion protein has
been suggested to play a pivotal role in ALK rearrangement-positive
IMT development, in a similar manner to that of the EML4-ALK fusion
protein in non-small cell lung cancer and that of NPM1-ALK or other
associated fusion proteins in aplastic large cell lymphomas
(20). However, considering that only
~50% of IMT cases present ALK gene rearrangement and that cases
with and without ALK gene rearrangement exhibit almost the same
clinical and pathohistological characteristics, it is possible that
unknown gene alterations may exist in these IMT cells. Although IMT
cells have slightly atypical cell morphology and are classified as
intermediate malignant tumors, aggressive local invasion or distant
metastasis are also observed. Further investigation of the
tumorigenicity of ALK rearrangement-negative IMT and establishment
of effective therapies for these tumors is required. In conclusion,
the current study reports a rare metastatic case of IMT in which
disease control was achieved through systemic chemotherapy.
References
|
1
|
Meis JM and Enzinger FM: Inflammatory
fibrosarcoma of the mesentery and retroperitoneum. A tumor closely
simulating inflammatory pseudotumor. Am J Surg Pathol.
15:1146–1156. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Coffin CM, Watterson J, Priest JR and
Dehner LP: Extrapulmonary inflammatory myofibroblastic tumor
(inflammatory pseudotumor). A clinicopathologic and
immunohistochemical study of 84 cases. Am J Surg Pathol.
19:859–872. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gleason BC and Hornick JL: Inflammatory
myofibroblastic tumors: Where we are now? J Clin Pathol.
61:428–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lawrence B, Perez-Atayde A, Hibbard MK,
Rubin BP, Dal Cin P, Pinkus JL, Pinkus GS, Xiao S, Yi ES, Fletcher
CD, et al: TPM3-ALK and TPM4-ALK oncogenes in inflammatory
myofibroblastic tumors. Am J Pathol. 157:377–384. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kovach SJ, Fischer AC, Katzman PJ, Salloum
RM, Ettinghausen SE, Madeb R and Koniaris LG: Inflammatory
myofibroblastic tumors. J Surg Oncol. 94:385–391. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
National Cancer Institute: Cancer Therapy
Evaluation Program: Common Toxicity Criteria. version 2.0.
March;1998.http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcv20_4-30-992.pdf
|
|
7
|
Eisenhauer EA, Therasse P, Bogaerts J,
Schwartz LH, Sargent D, et al: New response evaluation criteria in
solid tumours: Revised RECIST guideline (version 1.1). Eur J
Cancer. 45:228–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
U.S. Department of Health and Human
Services; National Institutes of Health. National Cancer Institute:
Common Terminology Criteria for Adverse Events (CTCAE). Version
4.0. May;2009.http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40
|
|
9
|
Lucas DR, Shukla A, Thomas DG, Patel RM,
Kubat AJ and McHugh JB: Dedifferentiated liposarcoma with
inflammatory myofibroblastic tumor-like features. Am J Surg Pathol.
34:844–851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Binh MB, Sastre-Garau X, Guillou L, de
Pinieux G, Terrier P, Lagacé R, Aurias A, Hostein I and Coindre JM:
MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing
well-differentiated and dedifferentiated liposarcoma subtypes: A
comparative analysis of 559 soft tissue neoplasms with genetic
data. Am J Surg Pathol. 29:1340–1347. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamamoto H, Oda Y, Saito T, Sakamoto A,
Miyajima K, Tamiya S and Tsuneyoshi M: p53 Mutation and MDM2
amplification in inflammatory myofibroblastic tumors.
Histopathology. 42:431–439. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Butrynski JE, D'Adamo DR, Hornick JL, Dal
Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ,
Ramaiya N, et al: Crizotinib in ALK-rearranged inflammatory
myofibroblastic tumor. N Engl J Med. 363:1727–1733. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bertocchini A, Lo Zupone C, Callea F,
Gennari F, Serra A, Monti L and de Ville de Goyet J: Unresectable
multifocal omental and peritoneal inflammatory myofibroblastic
tumor in a child: Revisiting the role of adjuvant therapy. J
Pediatr Surg. 46:E17–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Applebaum H, Kieran MW, Cripe TP, Coffin
CM, Collins MH, Kaipainen A, Laforme A and Shamberger RC: The
rationale for nonsteroidal anti-inflammatory drug therapy for
inflammatory myofibroblastic tumors: A children's oncology group
study. J Pediatr Surg. 40:999–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Przkora R, Bolder U, Schwarz S, Jauch KW,
Spes J, Andreesen R and Mackensen A: Regression of nonresectable
inflammatory myofibroblastic tumors after treatment with
nonsteroidal anti-inflammatory drugs. Eur J Clin Invest.
34:320–321. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kusunoki-Nakamoto F, Matsukawa T, Tanaka
M, Miyagawa T, Yamamoto T, Shimizu J, Ikemura M, Shibahara J and
Tsuji S: Successful treatment of an unresectable inflammatory
myofibroblastic tumor of the frontal bone using a cyclooxygenase-2
inhibitor and methotrexate. Intern Med. 52:623–628. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johnson K, Notrica DM, Carpentieri D,
Jaroszewski D and Henry MM: Successful treatment of recurrent
pediatric inflammatory myofibroblastic tumor in a single patient
with a novel chemotherapeutic regimen containing celecoxib. J
Pediatr Hematol Oncol. 35:414–416. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Favini F, Resti AG, Collini P, Casanova M,
Meazza C, Trecate G and Ferrari A: Inflammatory myofibroblastic
tumor of the conjunctiva: response to chemotherapy with low-dose
methotrexate and vinorelbine. Pediatr Blood Cancer. 54:483–485.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
ESMO/European Sarcoma Network Working
Group: Soft tissue and visceral sarcomas: ESMO Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol.
25:iii102–iii112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mano H: ALKoma: A cancer subtype with a
shared target. Cancer Discov. 6:495–502. 2012. View Article : Google Scholar
|