Introduction
Meningiomas are common tumors in the central nervous
system and have a large variety of histopathological appearances.
The majority of the variants are slow growing and follow a benign
clinical course without significant differences in prognosis.
However, certain subtypes of meningioma exhibit aggressive clinical
behavior, including atypical, chordoid, clear cell, papillary,
rhabdoid and anaplastic subtypes (1).
Chordoid meningiomas (CMs), which were recorded as a
novel subtype of meningioma in the 1993 World Health Organization
(WHO) classification of tumors (2),
are rare histological variants of meningioma that have been
reported to predominantly occur intracranially (3). CMs account for 0.5–1.0% of all
meningiomas and are more common in younger patients (<18 years).
These lesions demonstrate unique clinical associations and
prognostic implications (4).
Histopathologically, CM tumors are composed of epithelioid or
spindle cells arranged in cords or nests in a basophilic mucoid
matrix (2). Gross total resection is
the treatment of choice for CMs, however, in the event of residual
or recurrent CMs, postoperative radiotherapy is considered the
standard treatment (2,3). Although CM is considered a benign tumor
(WHO Grade II), this rare entity demonstrates a more aggressive
nature with a higher recurrence rate than that of typical
meningiomas (WHO Grade I). However, the overall prognosis of CM is
relatively good with a three-year survival rate of 93.4% (4,5).
Spinal meningiomas comprise almost 46% of all
primary spinal cord tumors, and they are extremely uncommon in
children (5). CM of the spinal canal
is also extremely rare, and to the best of our knowledge, there
have been only two cases of intraspinal CM reported in the
literature (6,7). The first case, reported by Couce et
al (6), was a 28-year-old woman
with a CM at the C2 level who underwent gross total resection of
the tumor and exhibited good recovery 5 years after surgery. The
second case, presented by Ibrahim et al (7), was that of a 26-year-old man who
harbored a C2–3 intradural and extramedullary chordoid meningioma.
The patient underwent gross total resection and exhibited no
neurological deficits 2 months after surgery.
The present study reports a case of spinal CM of the
cervical region in a 12-year-old girl. To the best of our
knowledge, the present study is the first case of pediatric spinal
CM to be reported in the literature. Written informed consent was
obtained from the patient's family.
Case report
A 12-year-old girl presented to the outpatient
clinic of Beijing Tiantan Hospital (Beijing, China) with a 3-month
history of progressive numbness and weakness in the right-side
limbs, and intermittent pain in the neck and right shoulder. The
patient did not experience any bowel or bladder symptoms.
Neurological examination revealed that muscle tone was increased in
the right lower limb, and muscle power was grade 4/5 in the right
upper and lower limbs, which was classified using the Medical
Research Council grading system (8).
Deep and superficial sensation in the right leg and superficial
sensation in the right arm were reduced. The right lower limb also
exhibited increased deep tendon reflexes and a positive Babinski
sign. Laboratory data was within the normal limits and the medical
history was unremarkable.
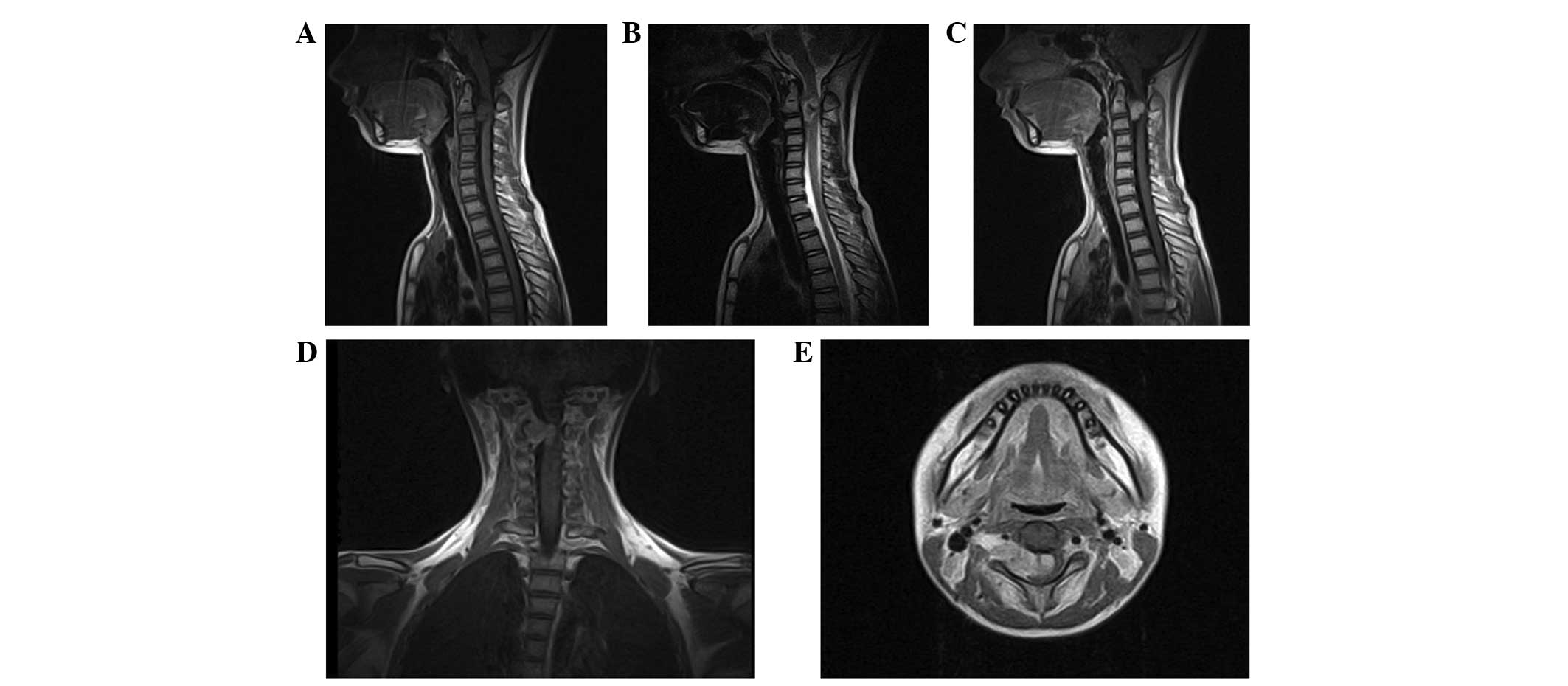
Pre-operative magnetic resonance imaging (MRI) of
the cervical spine demonstrated an intraspinal extramedullary
lesion at the C2–3 level on the right side of the spinal canal and
neural foramina, measuring 11×21×26 mm at the maximal dimensions
(Fig. 1). The tumor demonstrated
isointensity on T1-weighted images and mixed hyperintensity on
T2-weighted images. Contrast-enhanced MRI of the lesion revealed
irregularly heterogeneous enhancement subsequent to gadolinium
administration. The tumor severely compressed the spinal cord and
the cord was displaced to the right portion of the spinal canal.
The lesion extended through the enlarged C2-C3 neural foramen on
the right side as an exophytic paravertebral component, compressing
and partially encompassing the right vertebral artery. Abnormal
flow void signal, peritumoral edema and associated syringomyelia
were not observed. The brain MRI findings were normal. Other
imaging work-ups, including bone scanning and ultrasonography of
the abdomen, provided no novel information. According to the
location and MRI features of the tumor, the lesion was
pre-operatively diagnosed as schwannoma.
The patient underwent a C2-C3 laminotomy, which was
performed using the posterior midline approach. The dura was
extremely tense and a longitudinal incision was made in the center
of the dura. The intraoperative findings revealed that the tumor
possessed a complete capsule and was yellow in color, firm in
texture and moderately vascular. The tumor was firmly attached to
the ventrolateral surface of the dura. The spinal cord was
displaced to the left side of the thecal sac, and the spinal cord
was severely compressed. The expansile mass extended through the
right widening C2-C3 neural foramen and encompassed the adjacent
nerve rootlet. Due to the adhesion of the lesion to the dura, and
the proximity of the lesion to the right vertebral artery, a gross
total resection was challenging to achieve. The intradural contents
of the tumor were removed and, due to tumor infiltration, the
affected nerve rootlet was transected under spinal evoked-potential
monitoring, based on the protection of spinal functions. The
effects of cord compression discontinued, while the exophytic
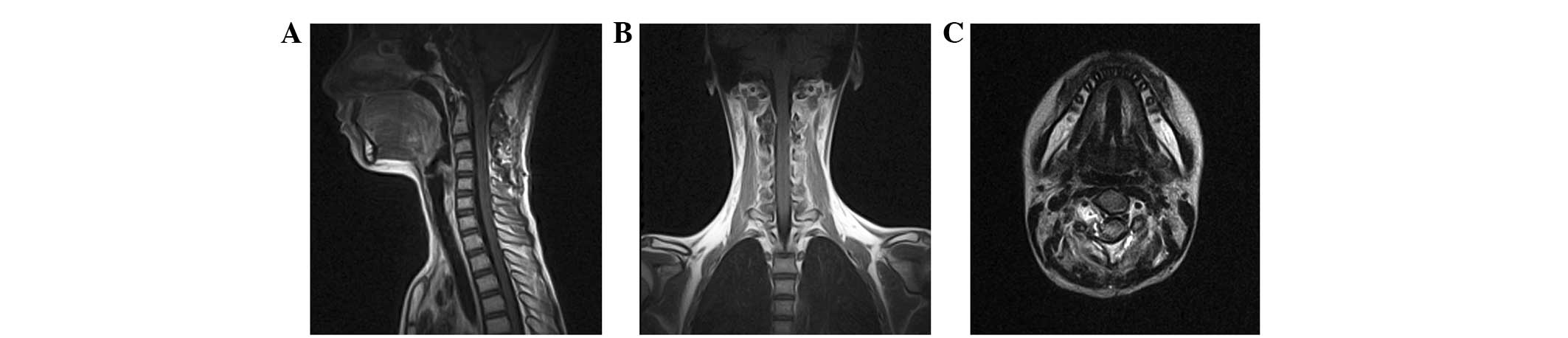
paravertebral component remained residual (Fig. 2).
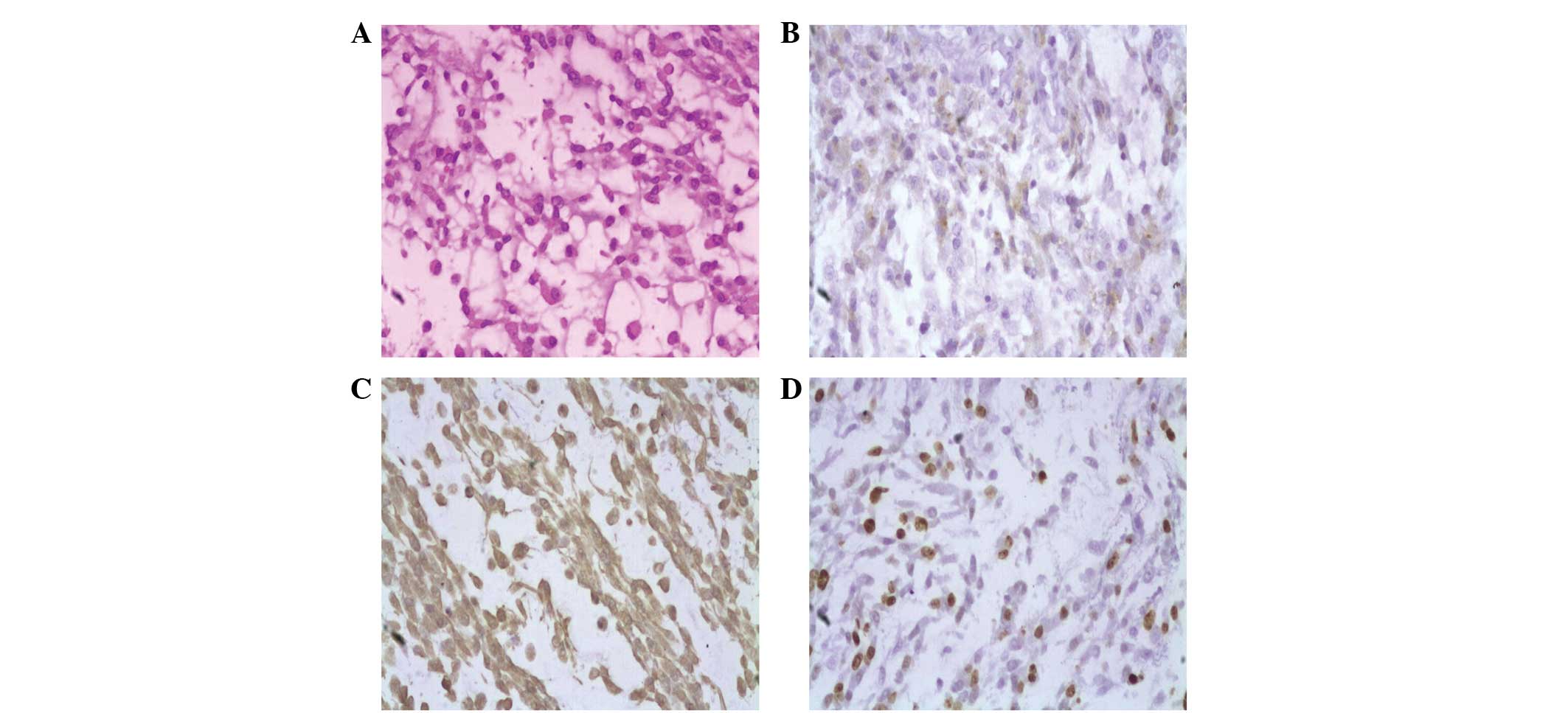
On histopathological examination, the tumor was
found to be composed of strands and cords of polygonal cells with
eosinophilic cytoplasm, several of which demonstrated vacuolation.
The majority of the tumor cells contained mucin-rich chordoid
elements embedded in the abundant myxoid matrix. The
immunohistochemical examinations revealed that the tumor cells were
positive for the expression of vimentin and epithelial membrane
antigen (EMA), but did not express desmin, S-100 or myogenin. In
total, ~20% of cells were positive for Ki-67. All these findings
were consistent with the diagnosis of CM (WHO Grade II) (Fig. 3).
The post-operative course was uneventful and the
sensory deficits were apparently relieved. Due to the aggressive
nature of lesions with high Ki-67 expression and the risk of tumor
recurrence, adjuvant radiotherapy was strongly recommended.
However, additional treatment for the residual spinal tumors was
refused and the patient was discharged from Beijing Tiantan
Hospital 2 weeks subsequent to the procedure. Gradual improvement
in the strength of the limbs was noted during the follow-up.
Regular follow-up MRI was performed, and the residual tumor
demonstrated no regrowth or metastasis 3 years subsequent to the
surgery. However, the patient experienced recurrence 5 years
subsequent to the procedure.
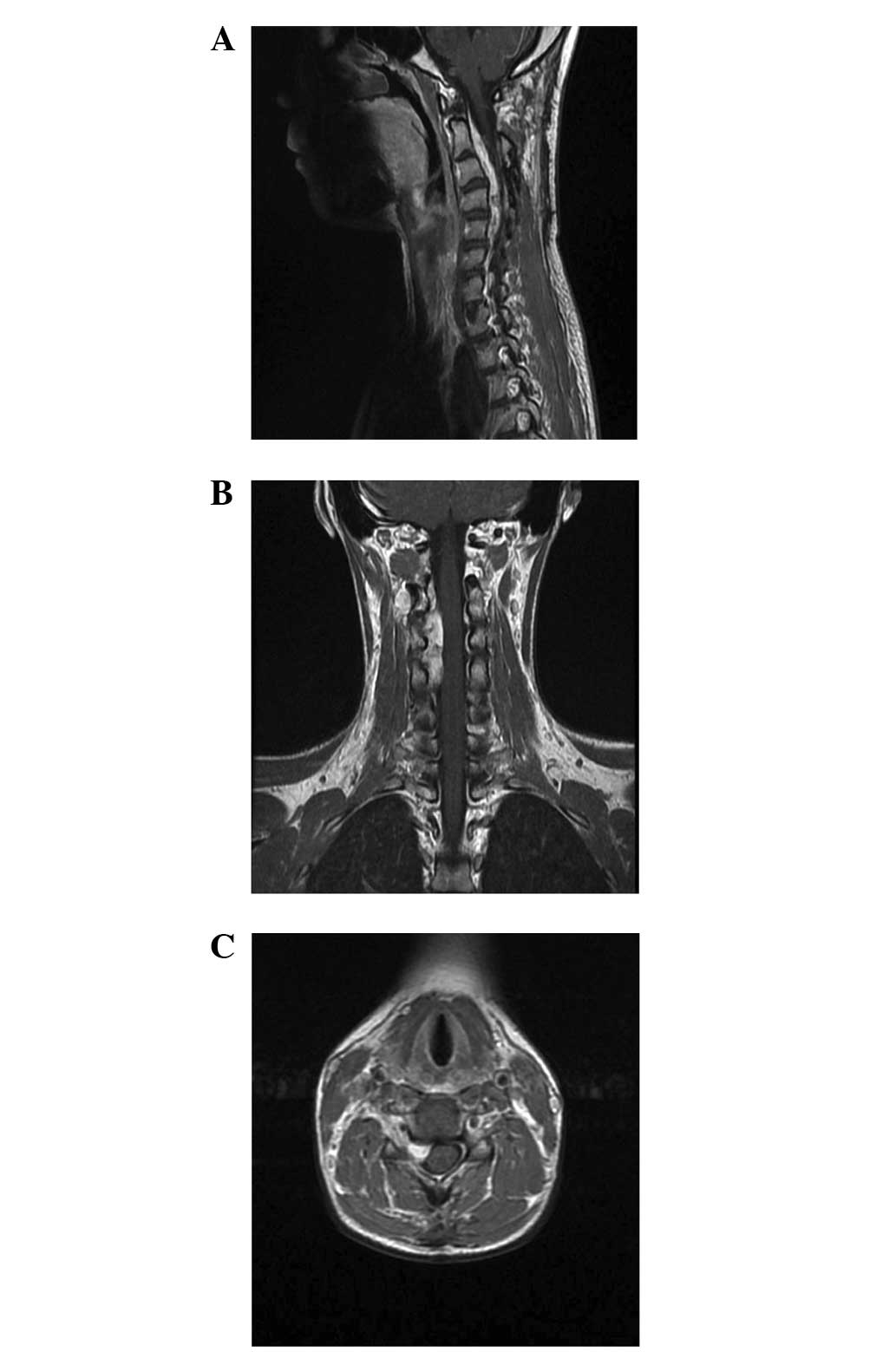
The patient presented with severe pain in the neck
and right shoulder that had recurred 2 months prior to admission to
the Department of Neurosurgery of Beijing Tiantan Hospital. An
immediate MRI revealed an extradural well-defined tumor that
demonstrated marked enhancement at the C2–5 level and involved the
right paraspinal region and adjacent neural foramen, measuring
70×30×17 mm at the maximal dimensions (Fig. 4). A C2–5 laminectomy was performed
using a posterior approach. The tumor was resected in sections and
partial removal was achieved using microsurgical techniques.
Histopathological examination confirmed the recurrence of CM.
Following the surgery, the patient received one course of radiation
therapy. External beam radiotherapy was administered to the
surgical site at a total dose of 40 Gy over 16 fractions. However,
the patient did not respond well to the treatment. The pain
worsened 1 month subsequent to surgery and the patient experienced
a progression of tetraplegia, with rapid enlargement of the
residual tumor. The patient succumbed to severe pneumonia and
respiratory failure 5 months subsequent to the second surgery.
Discussion
CM is a rare variant of meningioma that is
characterized by epithelioid cord-like tumor cells in the myxoid
stroma (4,9). CM has been recognized as an aggressive
tumor of meningothelial origin and is considered to be a grade II
lesion, in addition to clear cell and atypical meningioma, due to a
high rate of recurrence, particularly following subtotal resection.
Currently, >120 cases of CM have been reported in the
literature, and these cases account for 0.5–1.0% of all meningiomas
(5,10). The neuraxis may be involved at any
level with a significant predilection for the supratentorial
location and this rare entity occurs more commonly in young adult
patients in comparison with other subtypes of meningiomas. Compared
with typical meningiomas, the diagnosis and treatment of CMs is
critical for providing accurate information for the treatment and
prognosis of patients and establishing a reasonable schedule for
outpatient follow-up.
The occurrence of spinal CMs is exceedingly rare.
Since Couce et al described the first patient with spinal CM
in 2000, only two cases of spinal CM have been reported in the
English literature (6,7). Table I
summarizes the clinical features of the previously reported
patients and present patient with spinal CM. The age of all three
patients at the time of presentation was <30 years, which is
similar to the age of patients with intracranial CM, but different
from the age of patients with intraspinal typical meningiomas,
which mostly occur in patients 40–60 years old. All tumors arise
from the cervical spinal regions, while intraspinal meningiomas are
most frequently identified in the thoracic region of the spine. The
symptoms of CM, including somatic pain, numbness and weakness in
the limbs, are consistent with those of other common intraspinal
tumors, such as schwannoma and neurofibroma. In the present
patient, the duration between the onset of symptoms and
presentation was 3 months. This is shorter than the typical
duration for intraspinal meningiomas of 6 months, which is likely
to reflect the aggressive nature and relatively rapid growth
pattern of the tumor.
 | Table I.Summary of reported patients with
spinal chordoid meningiomas. |
Table I.
Summary of reported patients with
spinal chordoid meningiomas.
| Author, year
(Ref.) | Age, years | Gender | Duration of initial
symptoms | Tumor location | Symptoms | Treatment | Outcome |
|---|
| Couce et al,
2000 (6) | 28 | F | NA | C2 | NA | GTR | Good recovery at 5
years |
| Ibrahim et al,
2005 (7) | 26 | M | NA | C2–3, IDEM | Cramping and weakness
in lower limbs | GTR | Good recovery at 2
months |
| Present case | 12 | F | 3 months | C2–3, IDEM + ED | Numbness and weakness
of right-side limbs and intermittent pain in the neck and right
shoulder | 1st, PR; 2nd, PR +
RTX | Recurrence at 5
years; succumbed 5 months following the second surgery |
When first described, CM was hypothesized to affect
young patients that presented with one or more of the features of a
systemic inflammatory disorder associated with Castleman syndrome,
such as iron refractory microcytic anemia, hepatosplenomegaly,
retardation of somatic and sexual development, and bone marrow
dysfunction (4,11,12).
However, in certain studies (5,13), the
clinical manifestation of Castleman syndrome was not reported to be
a prerequisite for the diagnosis of CM, and the three reported
patients with spinal CM demonstrated no evidence of Castleman or
inflammatory syndrome.
MRI is the modality of choice for the diagnosis of
spinal tumors. Pre-operative differential diagnosis for intraspinal
tumors is important when planning surgical strategies and
determining the extent of the required resection to avoid
overtreatment and unacceptable complications. However, the imaging
features of CMs are indistinguishable from those of a typical
meningioma in the pre-operative period due to the lack of a highly
specific appearance. Thus, a definitive pre-operative diagnosis of
CM based only on MRI was challenging in the present study, and a
complete histopathological examination was required to
differentiate the CM from other intraspinal lesions.
Detailed pathological features of CMs have been
described in certain clinicopathological studies (4,5,7). Histologically, CM is characterized by
cords and clusters of spindle and epithelioid cells embedded in a
myxoid matrix, with vacuolated eosinophilic cytoplasm. The tumor
cells are usually diffusely positive for the expression of EMA and
vimentin. In certain cases of intracranial CM, the rapid
enlargement of the tumor over a relatively short time period may be
due to high mucin-producing activity (14). The histological differential diagnosis
of CM should include the following: chordoma; chondrosarcoma;
chordoid glioma; myxopapillary ependymoma; and clear cell
meningioma.
Due to the rarity of CM, the most effective method
of treatment remains ill-defined. When compared with typical
meningiomas (WHO grade I), which typically exhibit a good clinical
outcome and low risk of long-term recurrence following subtotal or
total resection, the presence of a WHO grade II meningioma should
indicate the requirement for a different management path, possibly
including adjuvant radiotherapy, closer follow-up and a lower
threshold for surgery in the event of recurrence. Since CM is
histologically benign and usually well circumscribed, gross total
removal using a microsurgical technique based on the protection of
the spinal function should be attempted during surgical
exploration. However, due to the adhesion of the tumor to the dura
and proximity to the vertebral artery, the tumor in the present
study was partially removed for mass effect relief to improve the
myelopathic symptoms and avoid severe operative complications. This
incomplete removal may have led to an increased risk of long-term
recurrence and caused a poorer outcome compared with the outcome of
the two previously reported cases, which were treated with total
removal.
It is hypothesized that adjuvant treatments, such as
radiotherapy or chemotherapy, may prevent tumor recurrence. In the
event of subtotally resected intracranial CMs, post-operative
radiotherapy is the standard treatment (10,14). Due
to the aggressive growth pattern of spinal CMs and long-term fatal
outcome in the present patient, post-operative early adjuvant
radiotherapy and close follow-up investigation in adult and
pediatric patients is recommended, although certain studies debate
the benefit of post-operative adjuvant treatments in pediatric
patients (2,7,15). The
efficacy of adjuvant radiation therapy for controlling spinal CMs
remains uncertain. In addition, other systemic therapies may be
considered for unresectable or recurrent tumors, but additional
investigation is required. Since the overall prognosis is uncertain
and long-term recurrence or regrowth is likely, regular follow-up
MRI is required.
The present study suggests that spinal CM should be
added to the differential diagnosis of intraspinal tumors in the
pediatric spine. Considering the aggressive nature of this rare
entity and the risk of long-term recurrence, total surgical removal
combined with adjuvant radiotherapy is recommended, although the
overall prognosis remains uncertain. Additional investigation with
long-term follow up and studies using larger sample sizes are
required.
Acknowledgements
The authors thank all physicians and staff that
provided aid in the present study.
References
|
1
|
Lantos PL, Van den Berg SR and Kleihues P:
Tumors of the nervous system. Greenfield's Neuropathology. Graham
DI and Lantos PL: 2:(6th). (London). Edward Arnold Publishers Ltd.
583–879. 1997.
|
|
2
|
Kleihues P, Burger PC and Scheithauer BW:
Histological typing of tumors of the central nervous system. World
Health Organization International Histological Classification of
Tumours (New York). Springer. 33–42. 1993.
|
|
3
|
Ozen O, Sar A, Atalay B, Altinörs N and
Demirhan B: Chordoid meningioma: Rare variant of meningioma.
Neuropathology. 24:243–247. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Tella OI Jr, Herculano MA, Prandini MN,
Stavile JN and Bonatelli Ade P: Chordoid meningioma: Report of two
cases. Arq Neuropsiquiatr. 61:91–94. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tena-Suck ML, Collado-Ortìz MA,
Salinas-Lara C, García-López R, Gelista N and Rembao-Bojorquez D:
Chordoid meningioma: A report of ten cases. J Neurooncol. 99:41–48.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Couce ME, Aker FV and Scheithauer BW:
Chordoid meningioma: A clinicopathologic study of 42 cases. Am J
Surg Pathol. 24:899–905. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ibrahim A, Galloway M, Leung C, Revesz T
and Crockard A: Cervical spine chordoid meningioma. Case report. J
Neurosurg Spine. 2:195–198. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Medical Research Council; Nerve Injuries
Research Committee: Aids to the investigation of peripheral nerve
injuries. His Majesty's Stationery Office (London). 481942.
|
|
9
|
Lin JW, Ho JT, Lin YJ and Wu YT: Chordoid
meningioma: A clinicopathologic study of 11 cases at a single
institution. J Neurooncol. 100:465–473. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Epari S, Sharma MC, Sarkar C, Garg A,
Gupta A and Mehta VS: Chordoid meningioma, an uncommon variant of
meningioma: A clinicopathologic study of 12 cases. J Neurooncol.
78:263–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Campos-Franco J, Otero E, Lopez-Garcia E,
Abdulkader I and Gonzalez-Quintela A: Chordoid meningioma in a
patient with lymphangioleiomyomatosis. J Neurooncol. 105:667–669.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaloshi G, Antonelli M, Vreto G, Lame A,
Kerri I, Bushati T, Rroji A and Petrela M: Report of two cases of
chordoid meningioma in patients with Castleman syndrome. J
Neurooncol. 104:395–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sangoi AR, Dulai MS, Beck AH, Brat DJ and
Vogel H: Distinguishing chordoid meningioma from their histologic
mimics: An immunohistochemical evaluation. Am J Surg Pathol.
33:669–681. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kano T, Nakazato Y, Tamura M, Ohye C, Zama
A, Saito F and Tomizawa S: Ultrastructural and immunohistochemical
study of an adult case of chordoid meningioma. Brain Tumor Pathol.
26:37–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Molleston MC, Moran CJ, Roth KA and Rich
KM: Infantile meningioma. Pediatr Neurosurg. 21:195–200. 1994.
View Article : Google Scholar : PubMed/NCBI
|