Introduction
Primitive neuroectodermal tumor (PNET) is a small
round cell sarcoma that primarily develops in the central nervous
system (CNS) and soft tissues of children (1). In particular, peripheral PNET (pPNET)
has been hypothesized to originate from the neural crest (2). Previously, PNET was suggested to be a
neoplasm of the central nervous system (3). Since then, this concept has been
extended to encompass the periphery, and these non-central nervous
system tumors are referred to as pPNETs (3). pPNET was initially described by Stout in
1918 (4). In addition, Ewing's
sarcoma (ES) of the bone was initially reported by J. Ewing in 1921
(5). Subsequent to this initial
description, a diagnosis of extraskeletal ES was reported by
Angervall and Enzinger in 1975 (6).
In 1979, Askin et al (7)
described a ‘malignant small cell tumor of the thoracopulmonary
region’ (Askin tumor). An additional significant discovery occurred
in 1984, when a neuroectodermal tumor of the bone was identified by
Jaffe et al (8). At the time
of diagnosis, these aforementioned tumors were recognized as being
pathologically distinct. However, it has since become apparent that
these tumors share a number of pathological features (7,8).
ES and pPNET (including Askin tumor) are proposed to
arise from the neural crest (9).
Currently, there are two opposing opinions that recognize ES and
pPNET as separate entities, or alternatively group them within a
single category known as the ‘ES/pPNET group’ (3) or the ‘ES family of tumors’ (10). Pathologically, when ES and pPNET are
recognized as separate entities, these distinctions are based
primarily on the more neural differentiation exhibited by pPNET
(11).
Typically, when considered from a histopathological
perspective, pPNETs possess similarities to a number of neoplasms,
including rhabdomyosarcoma and small cell carcinoma (4).
Within the head and neck region, the larynx is a
rare primary site for pPNET to arise (12). To the best of our knowledge, only a
small number of cases have previously been reported (13–15). In
these cases, follow-up periods were short and the details of
treatment were not discussed. The current report presents a case of
pPNET arising in the larynx, and subsequently reviews the
associated literature.
Case report
A previously healthy 33-year-old female presented
with the symptom of hoarseness ~6 months prior to admission. This
symptom was progressive, and the patient was unable to speak during
the initial consultation. Following clinical examination using a
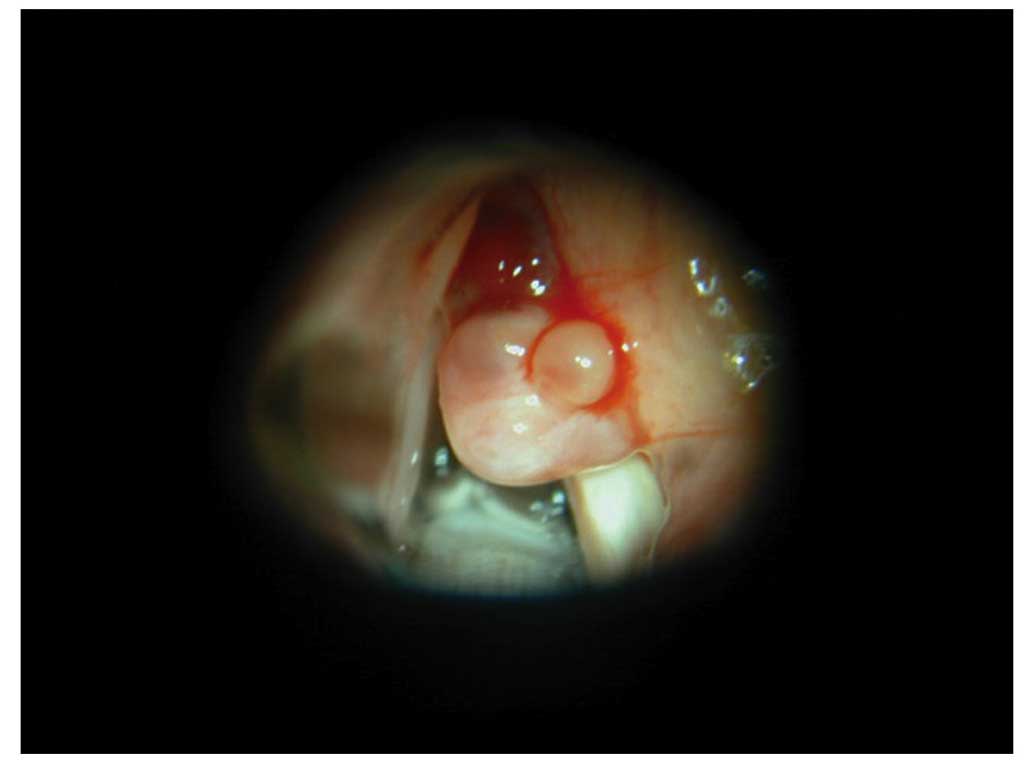
fiberscope, a slightly red tumor covered with white tissue was
identified on the right false vocal cord.
A punch biopsy was performed under fiberscopic view,
and the tumor was initially diagnosed as being neuroendocrine.
However, this diagnosis was unreliable due to the small size of the
biopsy specimen. In addition, the initial report indicated that the
tumor possessed low malignant potential. Computed tomography (CT)
and magnetic resonance imaging (MRI) studies were unable to detect
the tumor due to its small size.
Consequently, microscopic surgery was performed
under general anesthesia after admission to Nagoya Daini Red Cross
Hospital on June 28, 2001. The base of the tumor mass was
identified in the right laryngeal ventricle (Fig. 1), and dissected from the mucosa. The
tumor was resected, and subsequently a potassium titanyl phosphate
laser beam was applied to the tumor resection and the mucosa around
the tumor. A diagnosis of pPNET was reached following pathological
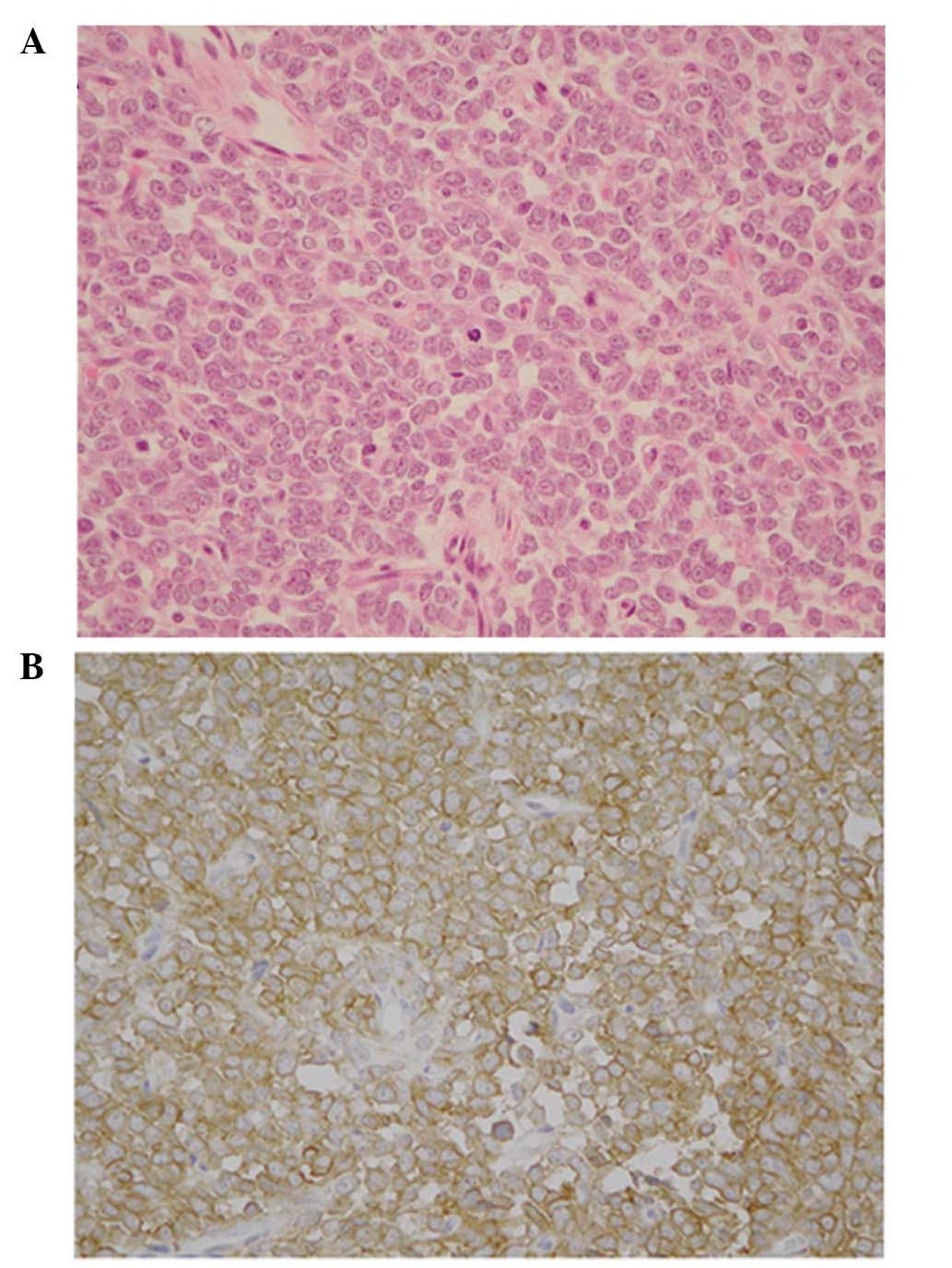
examination. Paraffin sections revealed sheets of small round
cells, which possessed relatively regular nuclei and sparse
cytoplasm. The nuclei demonstrated a salt and pepper chromatin
pattern, and the nucleoli were obscured. Abortive Homer-Wright
rosettes were rare, and no other specific patterns were identified.
A relatively high frequency of mitotic figures was observed, while
a Periodic Acid Schiff stain indicated that no glycogen was present
(Fig. 2A). Immunohistochemical
studies revealed that tumor cells were positive for cluster of
differentiation 99 (CD99), neuron-specific enolase (NSE), S100
protein and vimentin, and negative for cytokeratin (AE1/AE3,
CAM5.2), epithelial membrane antigen, muscle-specific actin,
α-smooth muscle actin, desmin, leucocyte common antigen,
chromogranin A and synaptophysin (Fig.
2B; Table I). An ultrastructual
study revealed that the tumor cytoplasm contained a reduced number
of mitochondria. Other organelles were not well developed; no
electron dense core granules were detected, and nuclei were round
with obscured nucleoli.
 | Table I.Immunohistochemical analysis
results. |
Table I.
Immunohistochemical analysis
results.
| Antibody | Result |
|---|
| Glycogen | − |
| Vimentin | + |
| S100 protein | + |
| Neuron-specific
enolase | + |
| Cluster of
differentiation 99 | + |
| Leucocyte common
antigen | − |
| Desmin | − |
| α-smooth actin | − |
| Synaptophysin | − |
| Muscle actin | − |
| Chromogranin | − |
Following diagnosis, CT scans of the chest, abdomen
and pelvis, an X-ray of all bones, an isotope bone scan, and an MRI
scan of the brain and bone marrow, were normal. Hematological tests
revealed a slight increase in the levels of lactate dehydrogenase
and creatine phosphokinase.
Additional surgery with a wide marginal resection,
including a total laryngectomy, was recommended; however, the
patient declined. The patient was therefore treated with multiagent
chemotherapy (ifosfamide, 50 µg/kg; vincristine, 2 µg/kg;
doxorubicin, 500 µg/kg). The patient also received radiotherapy of
a total dose of 60 Gy over 6 weeks.
The patient currently remains alive and healthy.
Five years subsequent to treatment, the patient has exhibited no
local disease recurrence or evidence of metastases.
Discussion
The concept of PNET has been the subject of
considerable discussion in multiple cases (8,9).
Initially, the term PNET was limited to tumors of the CNS. However,
non-CNS tumors possessing a similar histological origin have been
reported, and are known as pPNETs (3).
pPNET, with a primary site in the head and neck
region, is rarely diagnosed (12,16–18). The
most common primary site of pPNET is the chest wall, which is known
as Askin tumor. The second most common primary site of pPNET is the
pelvis (19,20). To the best of our knowledge, among the
associated literature, the present case is only the fourth report
of laryngeal pPNET (13–15). In addition, to the best of our
knowledge, in the 5 years following the presentation of this case,
no similar reports have been documented. Previous reported cases of
pPNET arising in the larynx are summarized in Table II (13–15).
| Table II.Review of reported cases of laryngeal
peripheral primitive neuroectodermal tumors and the present
case. |
Table II.
Review of reported cases of laryngeal
peripheral primitive neuroectodermal tumors and the present
case.
| Study | Year | Patient age,
years | Patient gender | Treatment | Metastases | Reference |
|---|
| Jones et
al | 1995 | 9 months | M | S+C | None | (13) |
| Yang et
al | 2004 | 74 | M | S+R | None | (14) |
| Lynch et
al | 2014 | 45 | F | C+R | None | (15) |
| Present case |
| 33 | F | S+C+R | None |
|
Histologically, typical pPNET is comprised of small
round, blue-stained cells possessing hyperchromatic nuclei, with a
high mitotic rate (21). The
cytoplasm is indistinct, except in regions where cells are more
mature, and the elongated hair-like cytoplasmic extensions coalesce
to form rosettes. The majority of the rosettes contain a central
solid core of neurofibrillary material known as a lobular or
pseudorosette, for example, the Homer-Wright rosette (13,22).
The ultrastructural characteristics of a typical
pPNET are primarily the presence of junctional complexes and
confluent cell processes, which contain occasional neurosecretory
type granules, microtubules and intermediate filaments (23).
Immunohistochemically, a number of authors have
reported that typical pPNETs are positive for NSE and S100 protein
staining, as well as additional markers, including Leu-7,
synaptophysin, neurofilament (24),
cytokeratin and chromogranin, which are positive at varying rates,
suggesting variation in neural differentiation (25). ES and pPNET apparently arise in
association with a translocation between chromosomes 11 and 22,
specifically t(11;22)(q24;q12). These two tumors also express
glycoprotein p30/32, coded for by the CD99 gene (HBA71 antigen or
MIC2), and are recognized by commercially available antibody, O13
(19).
Anti-CD99 antibodies have been recognized as
specific markers for the differential diagnosis of ES and pPNET, as
these tumors demonstrate strong positive expression of the CD99
gene (26). However, a number of
studies have reported that cases of bone and soft tissue tumors,
including T-lymphoblastic lymphoma (27), poorly differentiated synovial sarcoma
(28), small cell osteosarcoma
(29), rhabdomyosarcoma (26), desmoplastic small round cell tumor
(30), small cell carcinoma (31), Merkel cell carcinoma (32) and mesenchymal chondrosarcoma (33), also express CD99 gene products
(24–31).
Therefore, differential diagnoses of ES and pPNET
must be obtained from overall microscopic, ultrastructural and
immunohistochemical findings. In addition, identification of a
common cytogenetic abnormality t(11;22)(q24;q12) in these tumors
provides strong support for diagnosis.
A number of previous studies have reported that
pPNET is an aggressive disease, demonstrating high rates of local
recurrence and distant metastases at an early stage. The most
frequently observed sites of metastasis are the lungs and bone
(34). Numerous patients exhibiting
distant metastases at the time of diagnosis of pPNET possessed poor
prognoses (35). The treatment for
these pPNET cases was similar to that for rhabdomyosarcoma and
Ewing's sarcoma; however, a highly effective treatment strategy for
pPNETs remains to be established (36,37). In
previous years, the prognosis of pPNET was poor, but has improved
due to the administration of combined treatment strategies
consisting of surgery, radiotherapy and chemotherapy (34,38).
The range of surgical resection is controversial as
pPNET arises at a variety of sites, and the method of resection and
surgical margin is site-specific. For tumors located in the head
and neck, a wide surgical margin cannot be achieved. It has been
reported that when pPNETs were resected with a surgical margin
>10 cm, postoperative radiotherapy was not required (34). Regarding the larynx and with
consideration of the anatomy in the present study, it was
hypothesized that if the tumor did not invade the adjacent tissue,
including the pharynx and thyroid, laryngectomy was a wide enough
resection of pPNET, as the mucosa of the larynx was enclosed by
thyroid and cricoid cartilage. In the present case, the patient
refused a laryngectomy due to the fact that the resection was close
to the surgical margin and further surgery may have been required.
A number of previous studies have recommended resection of pPNETs,
with a wide surgical margin. It has been reported that patients who
underwent a resection with a wide surgical margin, demonstrated
improved overall survival, compared with those who underwent a
less-than-wide resection (39).
Additionally, for patients with localized tumors, wide resection
may facilitate favorable prognoses. Additional postoperative
radiotherapy may be capable of controlling local disease when
microscopic surgical margins remain pathologically positive
(34). The resection of all gross
pPNET tumors within 3 months of diagnosis is correlated with
significantly improved disease-free survival rates (20).
Furthermore, multiagent chemotherapy has been
recommended for the control of distant micrometastases by numerous
previous studies. Since the 1980s, preliminary reports have
suggested vincristine, ifosfamide, cyclophosphamide and doxorubicin
as chemotherapeutic agents for the treatment of metastasis of
Ewing's sarcoma. Identical chemotherapeutic strategies have been
performed on pPNETs, and the responses were similar to those
identified in Ewing's sarcoma, resulting in improved rates of
survival (10,35,38,40).
However, the optimal chemotherapeutic agents for the treatment of
pNET remain under investigation.
In previous studies, the prognosis of pPNET has been
demonstrated to be poor, although certain studies have demonstrated
improvements (34,39). Current literature indicates that the
overall 5-year survival rate is ~40–60%, whereas this figure was
48% subsequent to 1970, and 28% prior to this (34). In recent reports, 5-years survival
rate is around 60% (41–43). However, an improved prognosis has been
demonstrated by patients exhibiting pPNET arising in the head and
neck region, compared with those patients with tumors at
alternative sites (35,44,45). In a
previous study, 5 cases of pPNET were reported to have arisen at
head and neck sites. In these cases, an improved prognosis was
demonstrated, compared with tumors that arose at alternative sites,
and all 5 patients had survived 18 months later (46). This result suggested that complete
tumor resection with a wide margin, followed by local irradiation
and systemic chemotherapy may be concluded to provide an improved
prognosis for patients exhibiting pPNET, compared with that of
alternative treatment strategies.
In conclusion, pPNET in the head and neck region is
rare, particularly in the larynx. pPNET treatment strategies
consisting of combinations of surgery, chemotherapy and radiation
remain controversial. Further study is required in order to provide
clearer evidence regarding the optimal treatment strategy for
pPNET.
References
|
1
|
Riggi N and Stamenkovic I: The Biology of
Ewing sarcoma. Cancer Lett. 254:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
von Levetzow C, Jiang X, Gwye Y, et al:
Modeling initiation of Ewing sarcoma in human neural crest cells.
PLoS One. 6:e193052011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Batsakis JG, Mackay B and El-Naggar AK:
Ewing's sarcoma and peripheral primitive neuroectodermal tumor. An
interim report. Ann Otol Rhinol Laryngol. 105:838–843. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stout AP: A tumor of the ulnar nerve. Proc
NY Pathol Soc. 12:2–12. 1918.
|
|
5
|
Ewing J: Classics in oncology.
Histopathology. CA Cancer Clin J. 22:95–98. 1972. View Article : Google Scholar
|
|
6
|
Angervall L and Enzinger FM: Extraskeletal
neoplasm resembling Ewing's sarcoma. Cancer. 36:240–251. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Askin FB, Rosai J, Sibley RK, Dehner LP
and McAlister WH: Malignant small cell tumor of the
thoracopulmonary region in childhood, A distinctive
clinicopathologic entity of uncertain histogenesis. Cancer.
43:2438–2451. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaffe R, Santamaria M, Yunis EJ, Tannery
NH, Agostini RM Jr, Medina J and Goodman M: The neuroectodermal
tumor of bone. Am J Surg Pathol. 8:885–898. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dehner LP: Peripheral and central
primitive neuroectodermal tumors. A nosologic concept seeking a
consensus. Arch Pathol Lab Med. 110:997–1005. 1986.PubMed/NCBI
|
|
10
|
Wexler LH, DeLaney TF, Tsokos M, Avila N,
Steinberg SM, Weaver-McClure L, Jacobson J, Jarosinski P, Hijazi
YM, Balis FM and Horowitz ME: Ifosfamide and etoposide plus
vincristine, doxorubicin, and cyclophosphamide for newly diagnosed
Ewing's sarcoma family of tumors. Cancer. 78:901–911. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Furman J, Murphy WM, Jelsma PF, Garzotto
MG and Marsh RD: Primary primitive neuroectodermal tumor of the
kidney. Histopathology. Am J Clin Pathol. 106:339–344.
1996.PubMed/NCBI
|
|
12
|
Lane S, Ironside JW and Path MRC:
Extra-skeletal Ewing's sarcoma of the nasal fossa. J Laryngol Otol.
104:570–573. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jones JE and McGill T: Peripheral
primitive neuroectodermal tumors of the head and neck. Arch
Otolaryngol Head Neck Surg. 121:1392–1395. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang YS and Hong KH: Extraskeletal Ewing's
sarcoma of the larynx. J Laryngol Otol. 118:62–64. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lynch MC, Baker A, Drabick JJ, Williams N
and Goldenberg D: Extraskeletal Ewing's sarcoma arising in the
larynx. Head Neck Pathol. 8:225–228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chowdhury K, Manoukian JJ, Rochon L and
Begin LR: Extracranial primitive neuroectodermal tumor of the head
and neck. Arch Otolaryngol Head Neck Surg. 116:475–478. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Toda T, Atari E, Sadi AM, Kiyuna M and
Kojya S: Primitive neuroectodermal tumor in sinonasal region. Auris
Nasus Larynx. 26:83–90. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ferlito A: PrimaryE wing's sarcoma of the
maxilla: A clinicopathological study of four cases. J Laryngol
Otol. 92:1007–1024. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weidner N and Tjoe J: Immunohistochemical
profile of monoclonal antibody O13: Antibody that recognizes
glycoprotein p30/32MIC2 and is useful in diagnosing Ewing's sarcoma
and peripheral neuroepithelioma. Am J Surg Pathol. 18:486–494.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kushner BH, Hajdu SI, Gulati SC, Erlandson
RA, Exelby PR and Lieberman PH: Extracranial primitive
neuroectodermal tumors. Histopathology. Cancer. 67:1825–1829. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hart MN and Earle KM: Primitive
neuroectodermal tumors of the brain in children. Cancer.
32:890–897. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hachitanda Y, Tsuneyoshi M, Enjoji M,
Nakagawara A and Ikeda K: Congenital primitive neuroectodermal
tumor with epithelial and glial differentiation. Histopathology.
Arch Pathol Lab Med. 114:101–105. 1990.PubMed/NCBI
|
|
23
|
Mackay B, Luna MA and Butler JJ: Adult
neuroblastoma. Histopathology. Cancer. 37:1334–1351. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yao M, Dornfeld KJ, Buatti JM, Skwarchuk
M, Tan H, Nguyen T, Wacha J, Bayouth JE, Funk GF, Smith RB, et al:
Intensity-modulated radiation treatment for head-and-neck squamous
cell carcinoma - the University of Iowa experience. Int J Radiat
Oncol Biol Phys. 63:410–421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fellinger EJ, Garin-Chesa P, Glasser DB,
Huvos AG and Rettig WJ: Comparison of cell surface antigen HBA71
(p30/32MIC2), neuron-specific enolase, and vimentin in the
immunohistochemical analysis of Ewing's sarcoma of bone. Am J Surg
Pathol. 16:746–755. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ambros IM, Ambros PF, Strehl S, Kovar H,
Gadner H and Salzer-Kuntschik M: MIC2 is a specific marker for
Ewing's sarcoma and peripheral primitive neuroectodermal tumors.
Histopathology. Cancer. 67:1886–1893. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Riopel M, Dickman PS, Link MP and Perlman
EJ: MIC2 analysis in pediatric lymphomas and leukemias. Hum Pathol.
25:396–399. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gerald WL, Ladanyi M, de Alava E,
Cuatrecasas M, Kushner BH, LaQuaglia MP and Rosai J: Clinical,
pathologic, and molecular spectrum of tumors associated with
t(11;22)(p13;q12): Desmoplastic small round-cell tumor and its
variants. J Clin Oncol. 16:3028–3036. 1998.PubMed/NCBI
|
|
29
|
Devaney K, Vinh TN and Sweet DE: Small
cell osteosarcoma of bone, an immunohistochemical study with
differential diagnostic considerations. Hum Path. 24:1211–1225.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scotlandi K, Serra M, Manara MC, Benini S,
Sarti M, Maurici D, Lollini PL, Picci P, Bertoni F and Baldini N:
Immunostaining of the p30/32MIC2 antigen and molecular detection of
EWS rearrangements for the diagnosis of Ewing's sarcoma and
peripheral neuroectodermal tumor. Hum Pathol. 27:408–416. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lumadue JA, Askin FB and Perlman EJ: MIC2
analysis of small cell carcinoma. Am J Clin Pathol. 102:692–694.
1994.PubMed/NCBI
|
|
32
|
Perlman EJ, Lumadue JA, Hawkins AL, Cohen
K, Colombani P and Griffin CA: Primary cutaneous neuroendocrine
tumors. Histopathology. Cancer Genet Cytogenet. 82:30–34. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Granter SR, Renshaw AA, Fletcher CD, Bhan
AK and Rosenberg AE: CD99 reactivity in mesenchymal chondrosarcoma.
Hum Pathol. 27:1273–1276. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rud NP, Reiman HM, Pritchard DJ, Frassica
FJ and Smithson WA: Extraosseous Ewing's sarcoma. Histopathology.
Cancer. 64:1548–1553. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Raney RB, Asmar L, Newton WA Jr, Bagwell
C, Breneman JC, Crist W, Gehan EA, Webber B, Wharam M, Wiener ES,
et al: Ewing's sarcoma of soft tissues in childhood: A report from
the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol.
15:574–582. 1997.PubMed/NCBI
|
|
36
|
Harper PG, Pringle J and Souhami RL:
Neuroepithelioma - a rare malignant peripheral nerve tumor of
primitive origin, Report of two new cases and a review of the
literature. Cancer. 48:2282–2287. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hashimoto H, Enjoji M, Nakajima T, Kiryu H
and Daimaru Y: Malignant neuroepithelioma (peripheral
neuroblastoma). Histopathology. Am J Surg Pathol. 7:309–318. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Soule EH, Newton W Jr, Moon TE and Tefft
M: Extraskeletal Ewing's sarcoma: A preliminary review of 26 cases
encountered in the Intergroup Rhabdomyosarcoma Study. Cancer.
42:259–264. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ahmad R, Mayol BR, Davis M and Rougraff
BT: Extraskeletal Ewing's sarcoma. Cancer. 85:725–731. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kinsella TJ, Triche TJ, Dickman PS, Costa
J, Tepper JE and Glaubiger D: Extraskeletal Ewing's sarcoma,
Results of combined modality treatment. J Clin Oncol. 1:489–495.
1983.PubMed/NCBI
|
|
41
|
Gupta AA, Pappo A, Saunders N, Hopyan S,
Ferguson P, Wunder J, O'Sullivan B, Catton C, Greenberg M and
Blackstein M: Clinical outcome of children and adults with
localized Ewing sarcoma, Impact of chemotherapy dose and timing of
local therapy. Cancer. 116:3189–3194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rodríguez-Galindo CI, Liu T, Krasin MJ, Wu
J, Billups CA, Daw NC, Spunt SL, Rao BN, Santana VM and Navid F:
Analysis of prognostic factors in ewing sarcoma family of tumors,
Review of St. Histopathology. Cancer. 110:375–384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Smorenburg CH, van Groeningen CJ, Meijer
OW, Visser M and Boven E: Ewing's sarcoma and primitive
neuroectodermal tumour in adults, single-centre experience in The
Netherlands. Neth J Med. 65:132–136. 2007.PubMed/NCBI
|
|
44
|
Hafezi S, Seethala RR, Stelow EB, Mills
SE, Leong IT, MacDuff E, Hunt JL, Perez-Ordoñez B and Weinreb I:
Ewing's family of tumors of the sinonasal tract and maxillary bone.
Head Neck Pathol. 5:8–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kimber C, Michalski A, Spitz L and Pierro
A: Primitive neuroectodermal tumours, Anatomic location, extent of
surgery, and outcome. J Pediatr Surg. 33:39–41. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nikitakis NG, Salama AR, O'Malley BW Jr,
Ord RA and Papadimitriou JC: Malignant peripheral primitive
neuroectodermal tumor-peripheral neuroepithelioma of the head and
neck: A clinicopathologic study of five cases and review of the
literature. Head Neck. 25:488–498. 2003. View Article : Google Scholar : PubMed/NCBI
|