Introduction
Myeloproliferative neoplasms (MPNs) occur as a
result of continuous clonal proliferation of one or a multiple
series of mature and differentiated bone marrow cells (1). MPNs are characterized by an increased
number of bone marrow nucleated cells, terminal differentiation and
mature proliferative cells, and frequently occur without dysplasia
(2). These neoplasms usually exhibit
an insidious onset, with one or a multiple series of elevated
peripheral blood cells, often accompanied by hepatosplenomegaly.
Myelofibrosis, bone marrow failure and the development of acute
leukemia may also occur in the later stages of the disease
(3). Undifferentiated MPN accounts
for 10–20% of all MPN cases, and is characterized by
hepatosplenomegaly, leukocytosis, thrombocytosis and not having the
Philadelphia chromosome or breakpoint cluster region
protein-Abelson murine leukemia viral oncogene homolog 1 fusion
gene (4,5). Patients with undifferentiated MPN may or
may not have anemia or an elevated red blood cell count. These
characteristics do not conform to any diagnostic criteria;
therefore, it is challenging to diagnose.
The reported incidence of MPNs worldwide is ~0.1–3
cases per 100,000 individuals, with no significant differences
observed between regions and countries (6). The peak age of incidence is 50–60 years
old (6). Untreated MPN patients
exhibit a poor prognosis, and usually succumb within a few months
following diagnosis (7). However,
with the appropriate treatment certain patients may survive for a
number of years. The application of molecular targeted drugs for
MPN treatment may improve patient survival. Interferon (IFN)-α
specifically blocks the JAK2V617F mutation of hematopoietic stem
cells and may prevent the development of MPN (8). Previous studies concerning IFN-α
treatment of MPN patients demonstrate that IFN-α may completely
alleviate the hematology and molecular biology associated with MPN
patients (8,9).
The present study reported a case of a
myeloproliferative neoplasm (MPN) in a patient with a normal
complete blood cell count. The patient's condition was basically
stable. The present study also analyzed the diagnostic and clinical
features of MPNs, and a literature review was performed. MPN with a
normal complete blood cell count is a rare disease, and attention
should be focused on this entity in the clinic.
Case report
A 38-year-old female presented at Ningxia People's
Hospital (Yinchuan, Ningxia, China) in April 2013 with intermittent
splenomegaly, which was observed using ultrasound during an annual
health-check. A physical examination revealed an enlarged spleen,
but the liver was normal. Abdominal computed tomography also
identified splenomegaly. A gastroscopic examination revealed
gastric varices and multiple duodenal ulcers. Blood routine tests
showed a white blood cell count of 9.71×109/l (normal
range, 4.0–10.0×109/l), an absolute neutrophil count of
6.82×109/l (normal range, 2.0–7.0×109/l), a
monocyte count of 0.38×109/l (normal range,
0.1–0.6×109/l), an eosinophil count of
0.2×109/l (normal range, 0.02–0.52×109/l), a
red blood cell count of 4.5×1012/l (normal range,
3.5–5.5×1012/l), a hemoglobin level of 154 g/l (normal
range, 110–160 g/dl) and a platelet count of 208×109/l
(normal range, 100–300×109/l). Markers for hepatitis B
were negative and liver function tests were normal. Tests for tumor
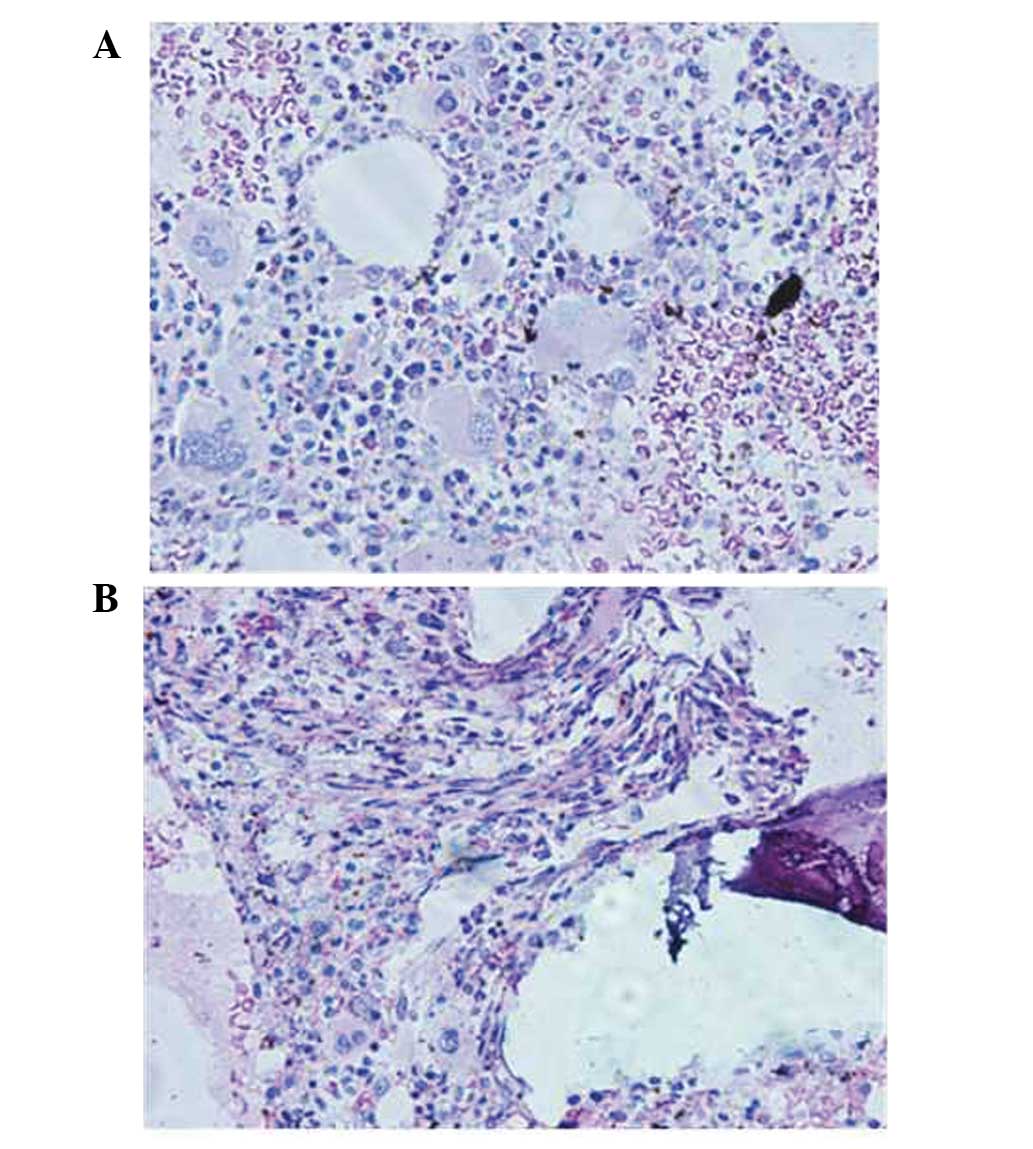
biomarkers were also normal. A bone marrow biopsy showed overactive
bone marrow hyperplasia, an elevated megakaryocyte count (clusters
of megakaryocytes were observed), megakaryocytic series dysplasia
and pleomorphic changes, multiple megakaryocyte clusters and focal
reticulin fiber hyperplasia (upon plastic embedding and hepatocyte
growth factor staining; Fig. 2). This
was indicative of a diagnosis of an MPN.
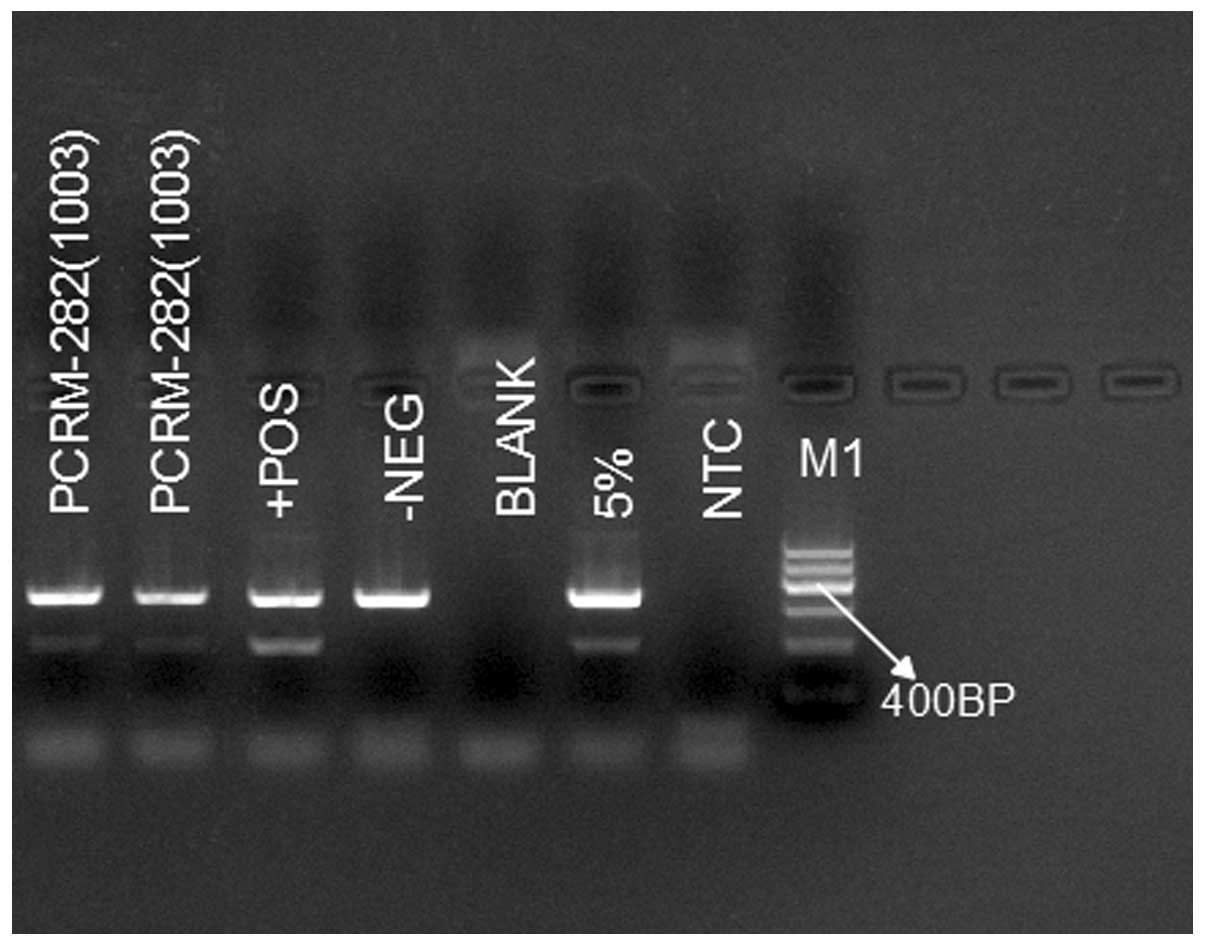
One week later, genetic analysis found the patient
to be positive for the JAK2-V617F mutation, and negative for the
JAK2 exon 12 and 13 mutations and the BCR-ABL (p210) fusion gene
(Fig. 1). T cell subgroup and
peripheral blood analyses were normal. The erythrocyte
sedimentation rate and autoantibody profile were also normal.
Repeat routine blood tests did not show any abnormalities. The
final diagnosis was of an unclassifiable MPN. No treatment was
administered and the patient was discharged with subsequent monthly
follow-up examinations planned. At the time of writing, the patient
was in a stable condition.
Discussion
MPNs, previously categorized under chronic
myeloproliferative disorders, are a type of hyperplastic disease of
stem cell origin, with heterogenous clinical features and
characterized by an increased peripheral blood cell count,
overactive bone marrow hyperplasia, and proliferation and
differentiation of mature hematopoietic cells (10). MPNs are neoplasms developed from the
continuous cloned proliferation of one or multiple series of
relatively mature and differentiated bone marrow cells,
characterized by increased bone marrow nucleated cells, terminal
differentiation and mature proliferative cells, and frequently
without dysplasia. MPNs usually have an insidious onset, with one
or multiple series of elevated peripheral blood cells, often
accompanied by hepatosplenomegaly. Chronic myelogenous MPNs include
leukemia, polycythemia vera (PV), essential thrombocythemia (ET),
chronic idiopathic myelofibrosis (CMF), chronic neutrophilic
leukemia, chronic eosinophilic granulocyte
leukemia/hypereosinophilic syndrome, and certain cases of
unclassifiable MPN (11–13). MF, bone marrow failure and the
development of acute leukemia can be observed in the later stage of
MPN. In 2005, multiple studies (11–15)
reported a high frequency of point mutations in the JAK2 gene,
JAK2-V617F, in cases of MPN and proposed that this mutation may be
a unique molecular biomarker of BCR/ABL-negative MPN. This finding
has brought about revolutionary progression in the research into
the pathogenesis, clinical classification, diagnosis and treatment
of MPNs, creating the so-called ‘JAK2 era.’ It has also been
reported that the incidence of the JAK2-V617F mutation in PV, ET
and CMF can be as high as 65–97, 23–57 and 35–57%, respectively
(16,17). However, the presence of the JAK2-V617F
mutation alone cannot be used to distinguish different categories
of MPN, but can be used for an accurate diagnosis when supplemented
with bone marrow cytology and bone marrow biopsy histology
(18).
Unclassifiable MPN is also termed as
undifferentiated MPN, accounting for 10–20% of MPN cases.
Unclassifiable MPN refers to diseases with clinical
(hepatosplenomegaly), laboratory (no Ph chromosome and/or BCR-ABL
fusion gene) and morphological (leukocytosis, thrombocytosis, with
or without anemia, with or without elevated red blood cell count)
characteristics of MPN, and does not conform to any diagnostic
criteria specific to MPN or include a disease overlap with >2
MPN characteristics. To the best of our knowledge, there have been
no previous MPN cases presenting with a normal complete blood cell
count. In the present patient, the complete blood cell count was
normal, and the patient presented with splenomegaly, tested
positive for the JAK2 gene and demonstrated bone marrow
pathological characteristics typical of MPN. Therefore, the patient
was diagnosed with unclassifiable MPN until confirmation could be
obtained. However, with no standard guidelines, this neoplasm could
not be classified. We propose that the patient's condition may be a
precursor of a certain type of MPN, and may later fall into one of
the established MPN categories, such as PV, ET or CMF.
The incidence of unclassifiable MPN is low, usually
with an occult onset and atypical manifestation. The disease
therefore requires special attention in order to avoid
misclassification, particularly in patients with normal complete
blood cell counts. However, the detection of JAK2 gene mutations
may aid the diagnosis, and thus enable the early detection and
treatment of unclassifiable MPN cases, and the prevention or
reduction in the incidence of thrombosis or hemorrhage. The
detection of JAK2 gene mutations is also of great significance in
the diagnosis, classification and effective management of MPNs.
In conclusion, MPN with a normal complete blood cell
count is rare. IFN-α may aid in the preventention of MPN
development; however, more clinical trials are required to confirm
this treatment. Recently, due to the identification of the JAK2,
MPL and CALR genes diagnosising MPN is easier (19). The case presented in the current study
is extremely rare; the clinical manifestation of the present
patient is atypical and easy to misdiagnosis. Therefore,
unexplained cases of splenomegaly with normal complete blood cell
counts should undergo routine bone marrow biopsies and JAK2 gene
testing in order to provide the correct diagnosis.
Acknowledgements
The authors would like to thank Professor Ling Su
(Ningxia People's Hospital) for collecting and managing the data
for the study.
References
|
1
|
Mahjoub S, Baccouche H, Sahnoun M, Kaabi
H, Manai Z, Slama H and Ben Romdhane N: The JAK2 mutation in
myeloproliferative neoplasms: A predictive factor of thrombosis.
Tunis Med. 93:474–477. 2015.PubMed/NCBI
|
|
2
|
Sørensen AL and Hasselbalch HC: Antecedent
cardiovascular disease and autoimmunity in Philadelphia-negative
chronicmyeloproliferative neoplasms. Leuk Res pii: S0145-2126.
30555(5)2015.(Epub ahead of print).
|
|
3
|
Hofmann I: Myeloproliferative neoplasms in
children. J Hematop. 8:143–157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abelsson J, Andréasson B, Samuelsson J,
Hultcrantz M, Ejerblad E, Johansson B, Emanuel R, Mesa R and
Johansson P: Patients with polycythemia vera have worst impairment
of quality of life among patients with newly diagnosed
myeloproliferative neoplasms. Leuk Lymphoma. 54:2226–2230. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shirane S, Araki M, Morishita S, Edahiro
Y, Ohsaka A and Komatsu N: Current problems in the diagnosis of
Philadelphia-negative myeloproliferative neoplasms in Japan. Rinsho
Ketsueki. 56:877–882. 2015.(In Japanese). PubMed/NCBI
|
|
6
|
Frederiksen H, Farkas DK, Christiansen CF,
Larsen TS, Hasselbalch HC, Stentoft J and Sørensen HT: Survival of
patients with chronic myeloproliferative neoplasms and new primary
cancers: A population-based cohort study. Lancet Haematol.
2:e289–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshida M, Horiuchi M, Ueda H, Hagihara K,
Kanashima H, Nakao T, Hirata C, Inoue T and Yamane T: Cutaneous
extramedullary hematopoiesis associated with
myelodysplastic/myeloproliferativeneoplasm, unclassifiable. Rinsho
Ketsueki. 56:911–914. 2015.(In Japanese). PubMed/NCBI
|
|
8
|
Kiladjian JJ: Current therapies and their
indications for the Philadelphia-negative
myeloproliferativeneoplasms. Am Soc Clin Oncol Educ Book.
2015:e389–396. 2015. View Article : Google Scholar
|
|
9
|
Schmitt-Graeff AH: Chronic myeloid
neoplasms. Diagnostic criteria and current therapeutic concepts.
Pathologe. 31:29–41. 2010.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tefferi A and Vardiman JW: Classification
and diagnosis of myeloproliferative neoplasms: The 2008 World
Health Organization criteria and point-of-care diagnostic
algorithms. Leukemia. 22:14–22. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baxter EJ, Scott LM, Campbell PJ, East C,
Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N,
et al: Acquired mutation of the tyrosine kinase JAK2 in human
myeloproliferative disorders. Lancet. 365:1054–1061. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jones AV, Kreil S, Zoi K, Waghorn K,
Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, et al:
Widespread occurrence of the JAK2 V617F mutation in chronic
myeloproliferative disorders. Blood. 106:2162–2168. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Levine RL, Wadleigh M, Cools J, Ebert BL,
Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et
al: Activating mutation in the tyrosine kinase JAK2 in polycythemia
vera, essential thrombocythemia and myeloid metaplasia with
myelofibrosis. Cancer Cell. 7:387–397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kralovics R, Passamonti F, Buser AS, Teo
SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M and Skoda RC: A
gain-of-function mutation of JAK2 in myeloproliferative disorders.
N Engl J Med. 352:1779–1790. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
James C, Ugo V, Le Couédic JP, Staerk J,
Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R,
Bennaceur-Griscelli A, et al: A unique clonal JAK2 mutation leading
to constitutive signalling causes polycythaemia vera. Nature.
434:1144–1148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levine RL and Wernig G: Role of JAK-STAT
signaling in the pathogenesis of myeloproliferative disorders.
Hematology Am Soc Hematol Educ Program. 233–239. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bumm TG, Elsea C, Corbin AS, Loriaux M,
Sherbenou D, Wood L, Deininger J, Silver RT, Druker BJ and
Deininger MW: Characterization of murine JAK2V617F-positive
myeloproliferative disease. Cancer Res. 66:11156–11165. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vardiman J, Brunning RD, Arber DA, et al:
Introduction and overview of the classification of the myeloid
neoplasms. WHO Classification of Tumours of Haematopoietic and
Lymphoid Tissues. Swerdlow SH, Campo E, Harris NL, et al: (Lyon,
France). IARC Press. 18–30. 2008.
|
|
19
|
Labastida-Mercado N, Galindo-Becerra S,
Garcés-Eisele J, Colunga-Pedraza P, Guzman-Olvera V, Reyes-Nuñez V,
Ruiz-Delgado GJ and Ruiz-Argüelles GJ: The mutation profile of
JAK2, MPL and CALR in Mexican patients with Philadelphia
chromosome-negative myeloproliferative neoplasms. Hematol Oncol
Stem Cell Ther. 8:16–21. 2015. View Article : Google Scholar : PubMed/NCBI
|