Introduction
Human hepatocellular carcinoma (HCC) is one of the
most widespread and severe types of malignancy in adults. An
estimated 782,500 new liver cancer cases and 745,500
cancer-associated mortalities occurred worldwide during 2012
(1). Certain treatments, including
liver transplantation, surgical resection, transcatheter arterial
chemoembolization, stereotactic body radiation therapy and
chemotherapy, are considered as the most effective therapies for
the treatment of HCC; however, these treatments are only effective
in certain patients (2). Therefore,
novel potential therapeutic targets are urgently required for the
management of HCC.
The Wnt/β-catenin signaling pathway has been
demonstrated to be dysregulated in HCC (3) and gastric (4), breast (5)
and colon cancers (6,7). It is one of the most significant
pathways that regulates cell proliferation, differentiation,
migration, apoptosis and survival. In normal epithelial cells,
β-catenin interacts and binds to the cytoplasmic tail of E-cadherin
and is sequestered to the membrane of the cell. In the cytoplasm of
the cell, a small amount of free β-catenin forms complexes with
glycogen synthase kinase 3β (GSK3β), adenomatous polyposis coli
(APC) and Axin. This complex is phosphorylated by Casein kinase 1
and GSK3β and is subsequently degraded by the
ubiquitination-proteasome pathway (8). When a Wnt ligand binds to a Frizzled
receptor (FZD), GSK3β is released from the GSK3β/APC/Axin complex,
leading to the activity of GSK3β becoming inhibited, and therefore
liberating β-catenin. β-catenin accumulates in the cytoplasm of the
cell and translocates to the nucleus where it regulates the
transcription of target genes through an interaction with the T
cell factor/lymphoid enhancer factor (TCF/LEF) family of
transcription factors and Legless family docking proteins (9,10).
The transforming growth factor-β (TGF-β) signaling
pathway has emerged as another key pathway in regulating tumor cell
growth and differentiation and maintaining the tumor interstitium
(11,12). In addition, TGF-β has been
demonstrated to act as an oncogenic cytokine by inducing
epithelial-mesenchymal transition (EMT), angiogenesis, and immune
suppression (13). When TGF-β binds
to a receptor complex composed of type I and type II
serine/threonine kinase receptors, signals are propagated to the
SMAD family of proteins, which activates the downstream signaling
molecules (14,15).
The synergistic activities of the TGF-β and
Wnt/β-catenin signaling pathways are pivotal during embryogenesis
due to their interaction with downstream effectors Smad 3 and 4 and
TCF/LEF (14,16). The effects of simultaneously targeting
TGF-β and Wnt/β-catenin signaling pathways in HCC has, to the best
of our knowledge, not been previously demonstrated. Consequently,
the present study investigated the synergistic antitumorous effects
caused by simultaneously blocking TGF-β and Wnt/β-catenin signaling
pathways using short hairpin (sh)-TGF-βRII and shFZD-7 in human HCC
HepG2 and Huh-7 cells.
Materials and methods
Cell culture
Human HCC HepG2 and Huh-7 cell lines were purchased
from the Cell Bank of Type Culture Collection of Chinese Academy of
Sciences (Shanghai, China) and were cultured in Gibco Dulbecco's
modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; GE
Healthcare Life Sciences, Logan, UT, USA), 100 U/ml penicillin and
100 µg/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA). All
cells were incubated at 37°C in an atmosphere of 5%
CO2.
shRNA synthesis and transfection into
pGPU6/GFP/Neo plasmids
shRNA sequences of TGF-βRII and FZD-7 were designed
by siRNA Sequence-Selector software (Vector NTI® version 10; Thermo
Fisher Scientific, Inc.). The oligonucleotide sequences were
inserted into BamHI and BbsI sites of pGPU6/GFP/Neo
plasmids (Shanghai GenePharma Co., Ltd., Shanghai, China). In
total, 8 shRNAs were designed to target 4 regions in the mRNA of
TGF-βRII and FZD-7. The 4 targeting sequences for TGF-βRII were as
follows: 5′-GCCCATCCACTGAGACATATT-3′, 5′-GGAGAAAGAATGACGAGAACA-3′,
5′-GCTTTGCTGAGGTCTATAAGG-3′ and 5′-GGAAGACAGAGAAGGACATCT-3′. The 4
targeting sequences for FZD-7 were as follows:
5′-GGTGGGTCATTCTGTCTCTCA-3′, 5′-GCCGTCAAGACCATCACTATC-3′,
5′-GTTCGTCTACCTCTTCATAGG-3′ and 5′-GCACCATCATGAAACACGACG-3′.
Therefore, a total of 8 plasmids (sh-TGF-βRII-a; sh-TGF-βRII-b;
sh-TGF-βR-c; sh-TGF-βRII-d; sh-FZD-7-1; sh-FZD-7-2; sh-FZD-7-3;
sh-FZD-7-4) were obtained for transfection. Scrambled shRNA that
did not cause specific degradation of any known cellular mRNA was
used as a negative control (sh-NC; sense,
5′-CACCGTTCTCCGAACGTGTCACGTCAAGAGATTACGTGACACGTTCGGAGAATTTTTTG-3′
and antisense,
5′-GATCCAAAAAATTCTCCGAACGTGTCACGTAATCTCTTGACGTGACACGTTCGGAGAAC-3′).
All the constructs were verified by sequence analysis.
shRNA transfection
The HCC cells were grown to 80% confluence prior to
transfection. sh-TGF-βRII, sh-FZD-7 and sh-NC plasmids were
transfected using Invitrogen Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. The
amounts of plasmid DNA in each transfection was equal (4 µg DNA and
10 µl Lipofectamine® 2000 in each group). The medium surrounding
the cells was replaced 4–6 h post-transfection to alleviate
toxicity.
Western blot analysis
The cells were homogenized in protein lysis buffer
(Beyotime Institute of Biotechnology, Nantong, Jiangsu, China), 48
h subsequent to transfection, and were centrifuged at 15,000 × g
for 15 min. The supernatant was extracted to obtain the total
cellular protein extracts. The concentration of the protein was
determined using the bicinchoninic acid assay (BCA kit; Beyotime
Institute of Biotechnology). The protein samples were denatured by
mixing with 5X loading buffer (Beyotime Institute of
Biotechnology). Following boiling for 5 min, the total cellular
protein extracts were separated using 8% sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) for β-catenin, 10%
SDS-PAGE for TGF-βRII, FZD-7, c-Myc and β-actin and 12% SDS-PAGE
for cyclin D1. The proteins were transferred onto nitrocellulose
membranes (GE Healthcare Life Sciences, Uppsala, Sweden) using a
wet or semi-dry transfer. The membranes were blocked with 5%
skimmed milk and Tris-buffered saline with Tween 20 (TBST) for 2 h
at room temperature, and rinsed three times with TBST for 30 min.
The proteins were incubated with primary polyclonal rabbit
anti-human antibodies against TGF-βRII, FZD-7, cyclin D1 and
β-actin (dilution, 1:1,000; bioWORLD, Dublin, OH, USA; catalog
no.'s., BS1696, BS2774, BS1741 and BS1002, respectively), and
monoclonal mouse anti-human antibodies against β-catenin and c-Myc
(dilution, 1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA; catalog no.'s., sc-59737 and sc-40, respectively). The
proteins were diluted with 0.5% skimmed milk in TBST overnight at
4°C, followed by rinsing three times with TBST for 30 min. The
proteins were incubated with the appropriate monoclonal goat
anti-mouse (or anti-rabbit) immunofluorescence-conjugated secondary
antibodies (dilution, 1:10,000; LI-COR Biotechnology, Lincoln, NE,
USA). The bands of specific proteins on the nitrocellulose
membranes were visualized with an Odyssey® CLx Infrared Imaging
System (LI-COR Biotechnology).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
The expression of TGF-βRII and FZD-7 mRNA in the
cells was assessed using RT-PCR to evaluate the efficiency of the
shRNA transfection. Total RNA from the transfected cells was
extracted using Trizol reagent (TianGen Biotech Co., Ltd., Beijing,
China), according to the manufacturer's protocol. In total, 1 µg of
RNA was reverse transcribed into cDNA using the Premium First
Strand cDNA Synthesis kit (TianGen Biotech Co., Ltd.). PCR primers
were ordered from Shanghai Genebase Gene-Tech Co., Ltd. (Shanghai,
China). The primer sequences obtained were as follows: GADPH sense,
5′-AGAAGGCTGGGGCTCATTTG-3′ and antisense,
5′-AGGGGCCATCCACAGTCTTC-3′, 258 bp; TGF-βRII sense,
5′-ATGCTGCTTCTCCAAAGTGC-3′ and antisense,
5′-AGTGCTCGCTGAACTCCAT-3′, 303 bp; FZD-7 sense,
5′-CTGTCGGGCTGCTACTTCAT-3′ and antisense,
5′-GCCAGGATAGTGATGGTCTTG-3′, 320 bp. The PCR reaction mixture
contained 1 µg template cDNA, 10 µM of each primer, 12.5 µl
2×Master Mix (TianGen Biotech Co., Ltd.) and 25 µl
ddH2O. The PCR amplification cycle was as follows:
Initial denaturation, 94°C for 5 min; 30 cycles consisting of 94°C
for 30 sec, 55°C for 30 sec and 72°C for 1 min; and storage, 72°C
for 5 min. An Eppendorf 5332 Mastercycler was used (Eppendorf,
Hamburg, Germany). PCR products were separated by electrophoresis
using 2% agarose gels.
Cell proliferation assay
The HepG2 and Huh-7 cells were incubated in 96-well
(5×103 cells/well) plates (Corning Inc., Corning, NY,
USA) in 100 µl medium. Subsequent to culturing for 24, 48, 72 and
96 h, the supernatant was removed and 100 µl serum-free DMEM and 10
µl cell counting kit-8 (CCK-8; Dojindo Molecular Technologies,
Inc., Kumamoto, Japan) solution was added to each well, followed by
incubation for 2 h at 37°C. The absorbance at 450 nm was recorded
using an ELX-800 Absorbance Reader (Bio-Tek Instruments, Inc.,
Winooski, VT, USA). All experiments were performed in quadruplicate
and repeated ≥3 times.
Clonogenic assay
For clonogenic analysis, the cells were harvested at
48 h post-transfection and ~3×103 cells from each
transfection group were seeded on 6-well culture plates. The cells
were incubated for 12–14 days and fixed with 4% paraformaldehyde
(Beijing Solarbio Sciences and Technology Co., Ltd., Beijing,
China) and stained with crystal violet (Sigma-Aldrich). The cell
colonies (≥50 cells) were counted using an Olympus microscope
(IX71+DP721; Olympus Corporation, Tokyo, Japan) at ×100
magnification.
Scratch wound healing assay
The cells were grown to confluence of 80–90% in
6-well culture plates to a density of 5×106 cells/well.
The confluent monolayer was disrupted with a 10-µl pipette tip and
washed three times with phosphate-buffered saline (PBS) to remove
cell debris. The wounded monolayer was photographed over the
following 24 h using a fluorescent microscope (Ti-U; Nikon
Corporation, Tokyo, Japan) at ×100 magnification. The migration
ability of the cells was determined by the ratio of the healing
width at 24 h to the wound width at 0 h.
Cell invasion and migration assay
Transwell chambers (EMD Millipore Corporation,
Billerica, MA, USA) containing an 8-µm pore polycarbonate membrane
filter was coated with 60 µl/well Matrigel (for an invasion assay;
BD Biosciences, San Jose, CA, USA) or without Matrigel (for a
migration assay) and inserted in a 24-well culture plate. The lower
chamber of the chamber was filled with 600 µl media supplemented
with 10% FBS as a chemoattractant, and 1×105 cells/well
in 200 µl serum-free DMEM were seeded into the upper chamber. The
chamber was incubated at 37°C in a humidified chamber in an
atmosphere of 5% CO2 for 24 h (migration assay) or 48 h
(invasion assay). Following incubation, the Transwell chambers were
removed. The cells in the upper chamber that did not migrate were
scraped away with a cotton swab. The cells that migrated through
and adhered to the lower chamber were fixed in paraformaldehyde for
30 min, then washed twice with PBS and stained with crystal violet
for 30 min. Images of the cells in the lower chamber were captured
using a fluorescence microscope (Ti-U; Nikon Corporation) equipped
with NIS-Elements F3.2. software (Nikon Corporation) at ×200
magnification.
Cell-cycle analysis
Single-cell suspensions containing 1×106
cells were collected at 48 h subsequent to transfection and fixed
with 70% ethanol for 2 h at 4°C. The cell cycle was monitored using
propidium iodide (PI; Nanjing KeyGen Biotec, Nanjing, China) to
stain the nuclei of the cells. The fluorescence of DNA-bound PI
cells was measured with a BD FACScan flow cytometer (FACSCanto™ II;
BD Biosciences), and the cell cycle distributions were analyzed
with ModFit LT™ version 3.0 software (Verity Software House, Inc.,
Topsham, ME, USA).
Statistical analysis
All experiments were conducted ≥3 times
independently. The experimental data were analyzed using SPSS
software version 16.0 (SPSS, Inc., Chicago, IL, USA) and
quantitative data were presented as the mean ± standard deviation.
Multiple groups were compared using one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Downregulation of TGF-βRII and FZD-7
expression by shRNA in human HCC HepG2 and Huh-7 cells
A total of 8 shRNAs were designed to target 4
regions in the mRNA of TGF-βRII and FZD-7. Following transfection,
the mRNA and protein levels of TGF-βRII and FZD-7 were examined by
RT-PCR and western blot analysis to screen which shRNA specifically
suppressed the expression of TGF-βRII and FZD-7. The mRNA and
protein levels of TGF-βRII and FZD-7 were significantly reduced in
cells transfected with sh-TGF-βRII and sh-FZD-7 plasmids compared
with the control groups (Fig. 1).
sh-TGF-βRII-c and sh-FZD-7-2 were demonstrated to be the most
effective specific suppressors. Transfection with sh-NC did not
affect TGF-βRII and FZD-7 expression at the mRNA or protein level
in HCC cells. Therefore, HepG2 and Huh-7 cells transfected with
sh-TGF-βRII-c and sh-FZD-7-2 plasmids were used for the following
analysis.
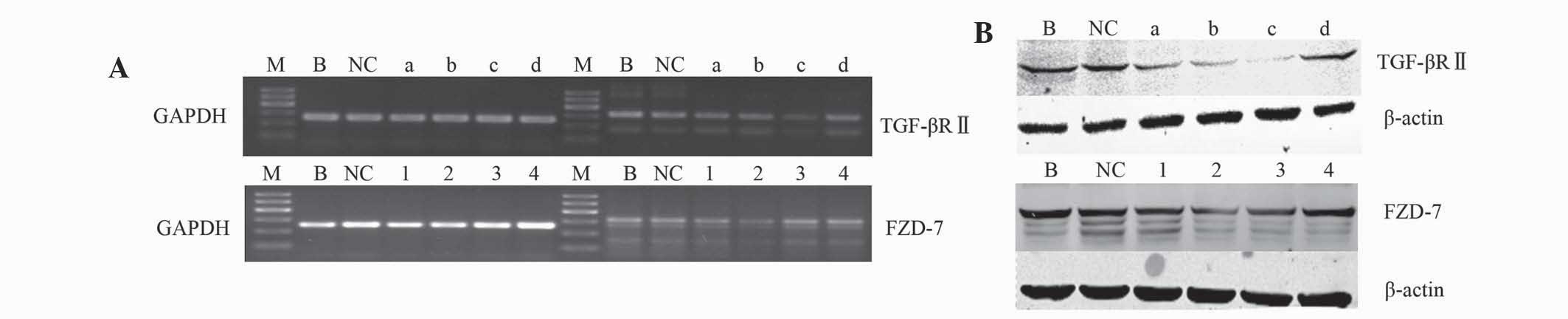 | Figure 1.(A) The mRNA and (B) protein levels of
TGF-βRII and FZD-7 in human hepatocellular carcinoma cells. GADPH
and β-actin were used as the loading controls. Reverse
transcription-polymerase chain reaction and western blot analysis
results were obtained from 3 experiments. TGF-β, transforming
growth factor-β; TGF-βRII, tranforming growth factor receptor II;
FZD, frizzled receptor; NC, negative control; shRNA, short hairpin
RNA; M, DNA marker; B, blank; NC, sh-NC; a, sh-TGF-βRII-a; b,
sh-TGF-βRII-b; c, sh-TGF-βRII-c; d, sh-TGF-βRII-d; 1, sh-FZD-7-1;
2, sh-FZD-7-2; 3, sh-FZD-7-3; 4, sh-FZD-7–4. |
Silencing of TGF-βRII and FZD-7
inhibited HCC cell proliferation
CCK-8 assays were used to determine if silencing
TGF-βRII and FZD-7 affects the growth of HCC cells. As demonstrated
by Fig. 2A, the proliferation of
transfected cells was significantly suppressed at various time
points (48, 72 and 96 h), particularly in the co-transfection group
(sh-TGF-βRII and sh-FZD-7) compared with cells in the
single-transfection group (sh-TGF-βRII or sh-FZD-7) at 96 h
(P<0.05). However, no significant differences were observed
between the blank and sh-NC groups (P>0.1). The proliferation
ability of the HCC cells was also assessed using colony formation
assays; the results were consistent with those of the CCK-8 assays
(Fig. 2B).
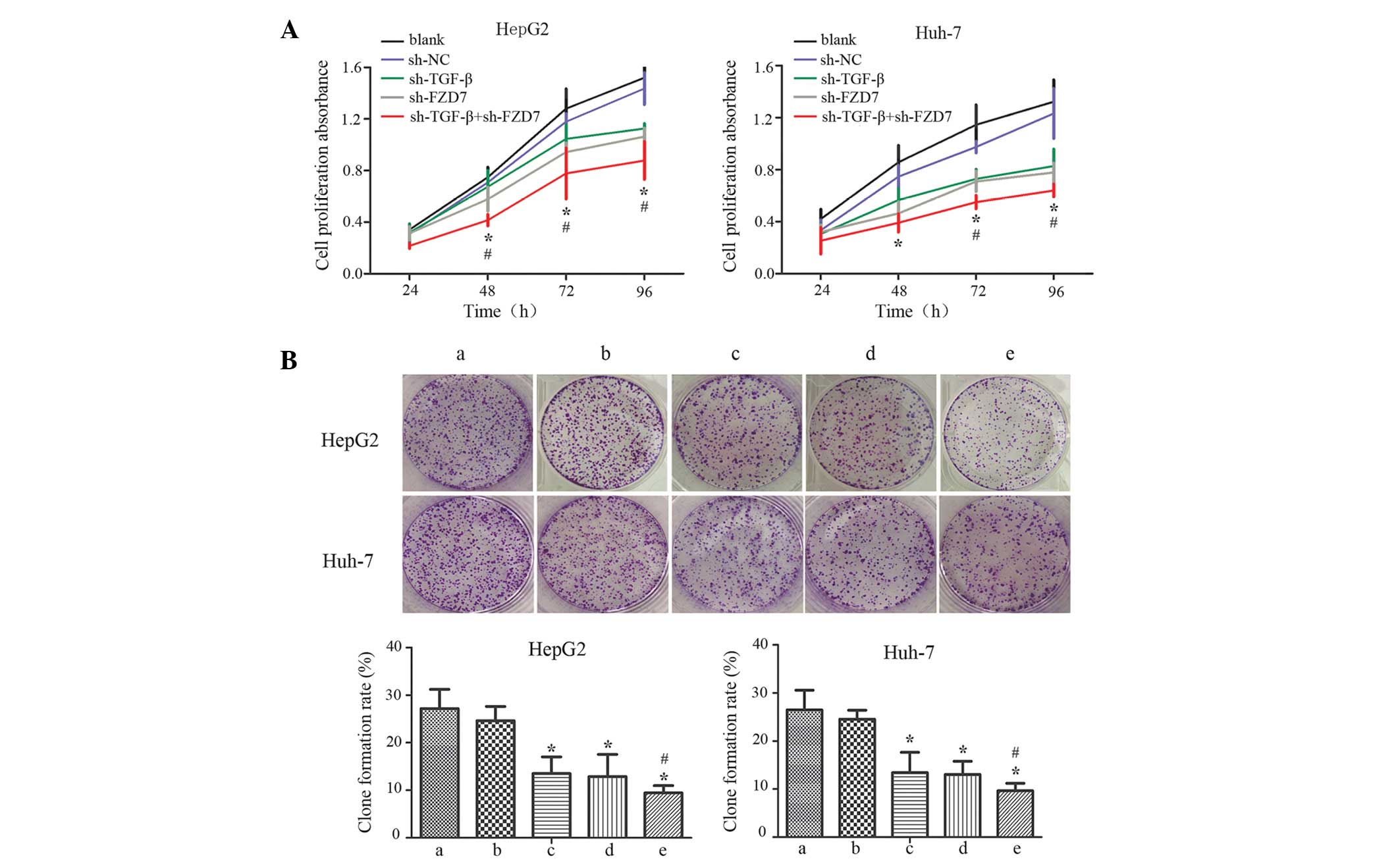 | Figure 2.Anti-proliferation effects of
sh-TGF-βRII and sh-FZD-7 on HCC cells . (A) A cell counting assay
was performed to examine the anti-proliferative effects of the
co-transfection of sh-TGF-βRII and sh-FZD-7 in HCC HepG2 and Huh-7
cells. *P<0.01 and #P<0.05. (B) The colony-forming
abilities of the cells were downregulated in the co-transfection
group compared with the single-transfection group in HepG2 and
Huh-7 cells. Each bar represents three independent experiments
exhibited as the mean ± standard deviation; error bars represent
the standard deviation. *P<0.01 compared with a and b;
#P<0.05 compared with c and d. TGF-βRII, transforming
growth factor-β receptor II; FZD, frizzled receptor; shRNA, short
hairpin RNA; HCC, human hepatocellular carcinoma; NC, negative
control; a, blank; b, sh-NC; c, sh-TGF-βRII; d, sh-FZD-7; e,
sh-TGF-βRII + sh-FZD-7. |
Cellular metastasis was impaired in
HCC cells transfected with sh-TGF-βRII and sh-FZD-7
Cellular migration and invasion are two essential
processes in cancer development. Therefore, the present study
investigated the cellular metastasis of HCC to demonstrate the
potential roles of TGF-β and Wnt/β-catenin signaling. The wound
healing assay revealed that the wound was healed more slowly when
cells were transfected with sh-TGF-βRII and sh-FZD-7 compared with
the control cells (Fig. 3A). The cell
migration assay demonstrated that co-transfection reduced the cell
migration ability by 67 and 68% for HepG2 cells and by 23 and 29%
for Huh-7 cells compared with the sh-TGF-βRII and sh-FZD-7 groups
(Fig. 3B). Additionally, invasion
assays were used to measure the invasion ability of the cells
through filters coated with reconstituted basement membrane. The
results demonstrated that simultaneous silencing of TGF-βRII and
FZD-7 inhibited the invasive ability of the cell by 41 and 43% for
HepG2 cells and by 41 and 38% for Huh-7 cells compared with the
sh-TGF-βRII and sh-FZD-7 groups (Fig.
3C).
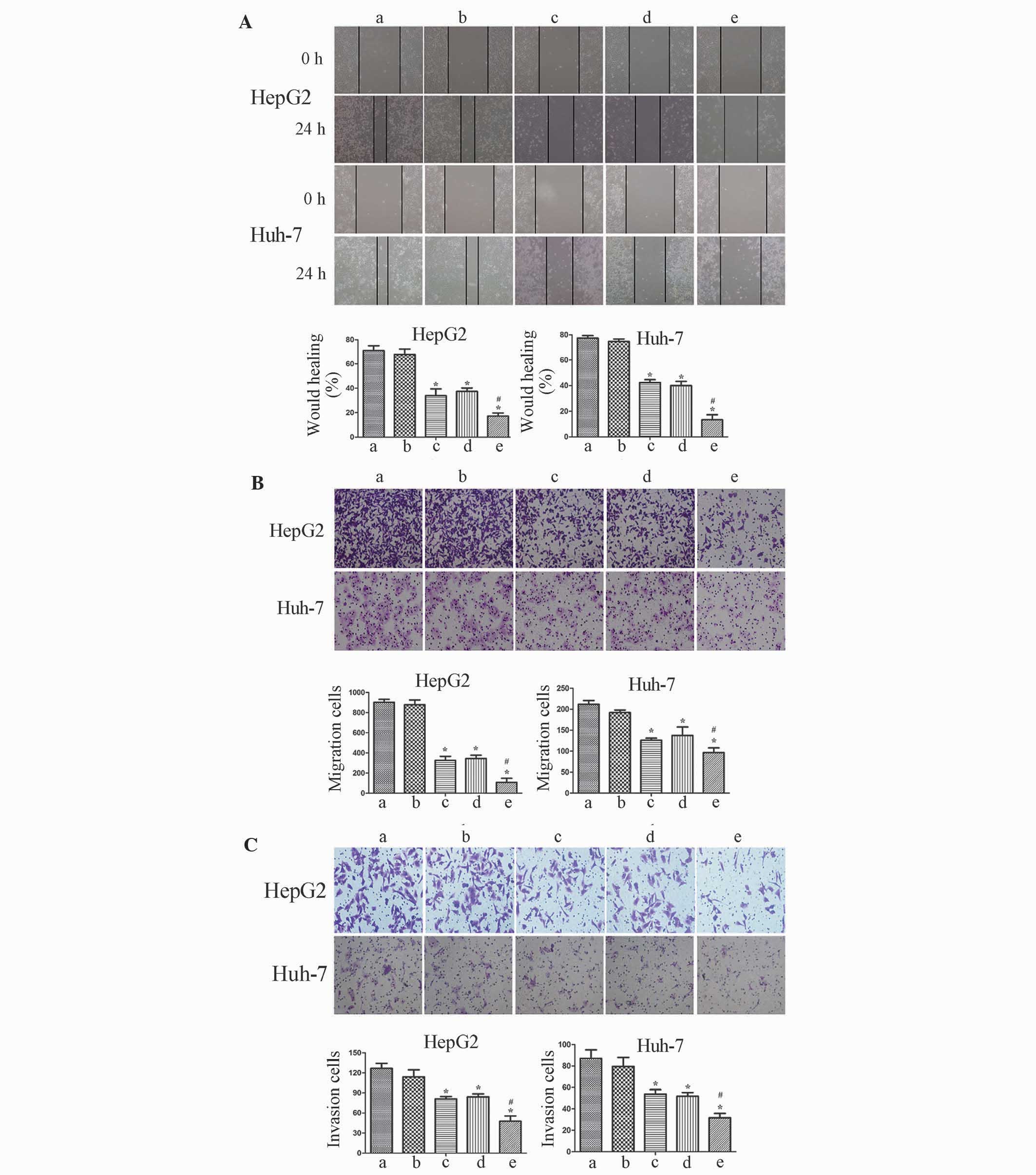 | Figure 3.Transforming growth factor-β and
Wnt/β-catenin signaling pathways regulate the migration and
invasion of human hepatocellular carcinoma cells. (A) A wound
healing assay was performed following TGF-βRII and FZD-7 knockdown
in HepG2 and Huh-7 cells. (B) A cell migration assay was performed
following TGF-βRII and FZD-7 knockdown in HepG2 and Huh-7 cells.
(C) Matrigel cell invasion abilities were downregulated in the
co-transfection group compared with the single-transfection group
in HepG2 and Huh-7 cells. Images of the cells that migrated onto
the lower chamber of a Transwell plate were captured under a light
microscope, at ×200 magnification. Each bar represents three
independent experiments presented as the mean ± standard deviation;
error bars represent the standard deviation. *P<0.01 compared
with a and b; #P<0.05 compared with c and d.
TGF-βRII, tranforming growth factor receptor II; FZD, frizzled
receptor; NC, negative control; sh, short hairpin RNA; a, blank; b,
sh-NC; c, sh-TGF-βRII; d, sh-FZD-7; e, sh-TGF-βRII + sh-FZD-7. |
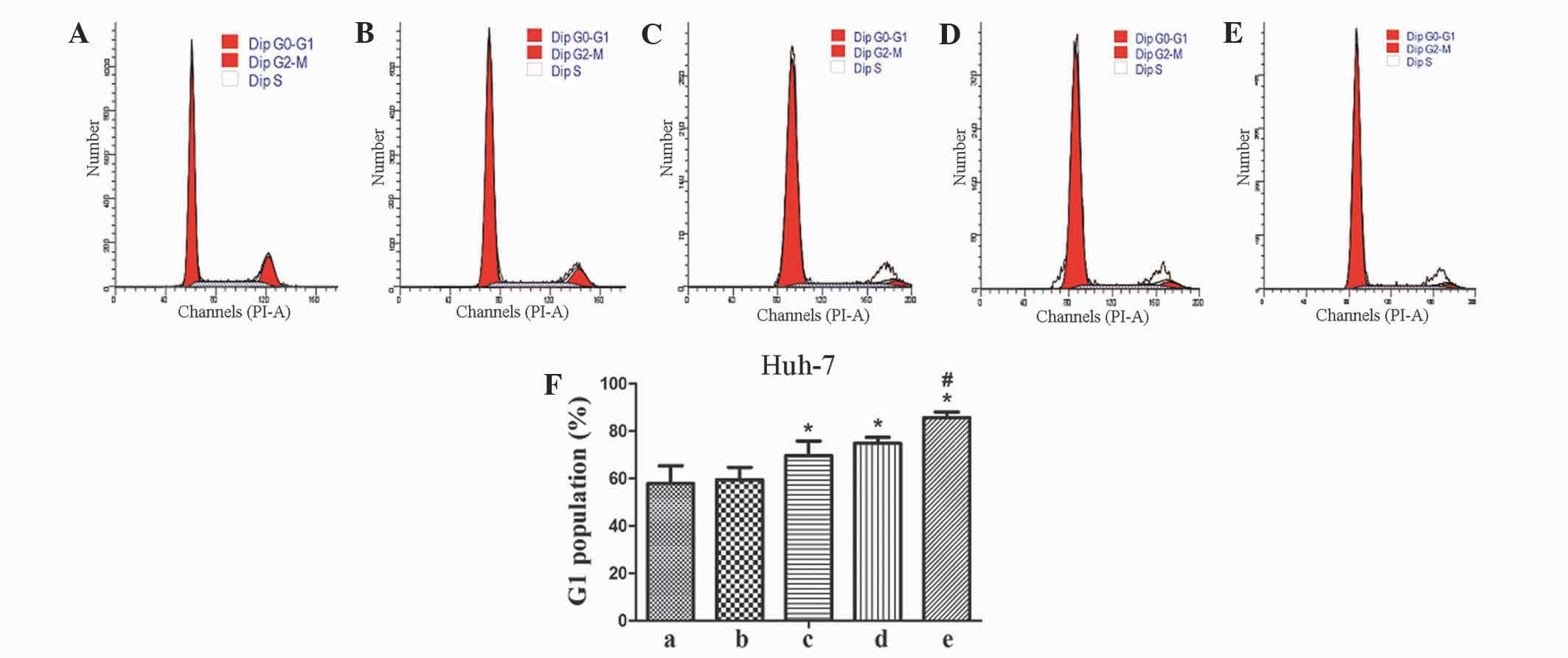
Cells arrest at the G1 phase, which
contributes to the reduction in cell proliferation
To investigate the mechanisms involved in the
reduction of cell proliferation in TGF-βRII and FZD-7 knockdown
cells, the present study performed flow cytometric analysis. As
demonstrated in Fig. 4, TGF-βRII and
FZD-7 knockdown cells led to a 18 and 13% increase in the
percentage of cells in G0-G1 phase compared with the sh-TGF-βRII
and sh-FZD-7 groups for Huh-7 cells. However, a statistical
difference was not observed between control and knockdown groups in
the percentage of HepG2 cells in G0-G1 phase (data not shown).
The expression of β-catenin, c-Myc and
cyclin D1 are downregulated in knockdown TGF-βRII and FZD-7 HCC
cells
In order to investigate the possible underlying
mechanisms of reduced proliferation and migration of HCC cells
co-transfected with sh-TGF-βRII and sh-FZD-7, the protein
expression levels of correlative genes, including β-catenin, c-Myc
and cyclin D1, were analyzed. Western blot analysis demonstrated
that the β-catenin, c-Myc, cyclin D1 protein levels were markedly
reduced in the co-transfection group compared to the
single-transfection groups (Fig.
5).
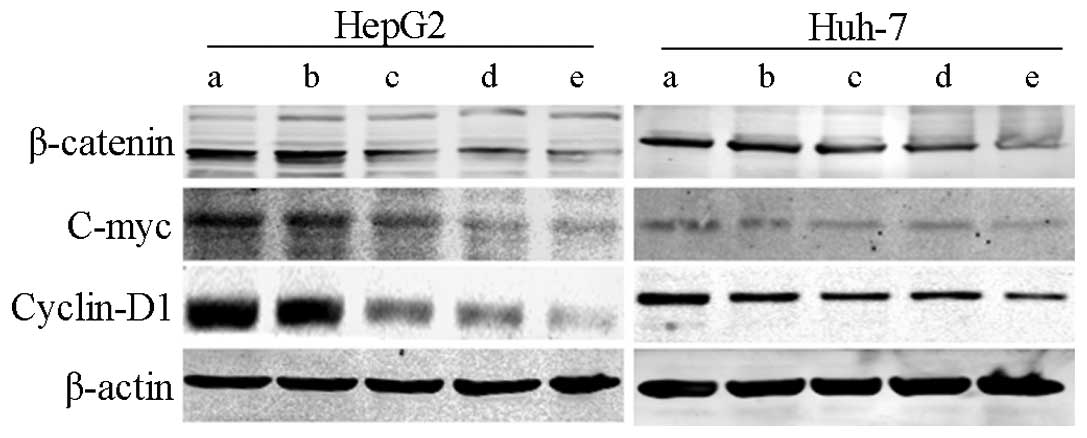 | Figure 5.Knockdown of TGF-βRII and FZD-7
suppresses β-catenin, c-Myc and cyclin D1 expression in human
hepatocellular carcinoma HepG2 and Huh-7 cells. β-actin was probed
as a loading control. TGF-βRII, tranforming growth factor receptor
II; FZD, frizzled receptor; NC, negative control; shRNA, short
hairpin RNA; a, blank; b, sh-NC; c, sh-TGF-βRII; d, sh-FZD-7; e,
sh-TGF-βRII + sh-FZD-7. |
Discussion
TGF-β and Wnt/β-catenin signals composed of two
groups of secreted proteins that act simultaneously to stimulate
the development of hepatopathy and HCC (17). Accumulating evidence has revealed that
TGF-β plays a dual role in cancer progression. At the early stages
of tumorigenesis, particularly when the tumor is benign, TGF-β acts
directly on the cell to suppress the tumor growth (11). However, as the tumor develops, genetic
or biochemical alterations allow TGF-β to stimulate tumor
progression in the cancer cell and surrounding non-malignant
stromal cells. The stimulation of the migration and invasion
abilities of cells by TGF-β may be of a greater clinical
consequence compared to its tumor-suppressive role, since the
majority of tumors retain functional TGF-β signaling pathways
(18). In addition, β-catenin is an
essential component of two cellular pathways; maintaining
cell-to-cell adhesion and mediating the Wnt/β-catenin signal
transduction pathway, which is pivotal in embryogenesis and
malignant transformation of cells (19).
The Wnt receptor complex on the cell surface
consists of a 7 transmembrane domain FZD protein and a single-pass
transmembrane protein from the low density lipoprotein
receptor-associated protein family. The activation of the Wnt
receptor complex results in the stabilization of cytoplasmic
β-catenin (8). Merle et al
(20) reported that the
overexpression of FZD-7 was detected in 90% of HCC cells, the
majority of which were associated with chronic hepatitis B virus
infection. In addition, a functional analysis demonstrated that the
levels of FZD-7 mRNA was associated with enhanced cellular
motility. By contrast, the TGF-β signaling pathway exerts its
various effects through two transmembrane serine/threonine kinases,
termed type I and type II receptors. The ligand-activated type II
receptor associates, phosphorylates and activates the type I
receptor, which in turn phosphorylates the members of the SMAD
protein family (21). These findings
indicate that FZD-7 and TGF-βRII are key gene targets for
interfering with Wnt/β-catenin and TGF-β signaling pathways.
In the present study, pGPU6/GFP/Neo coding plasmids
containing shRNA targeting TGF-βRII and FZD-7 were constructed to
investigate the effects of simultaneously blocking TGF-β and
Wnt/β-catenin signaling pathways in HCC cells. The expression
levels of TGF-βRII and FZD-7 were determined by RT-PCR and western
blot analysis. The present study demonstrated that sh-TGF-βRII-c
and sh-FZD-7-2 significantly downregulated the expression of
TGF-βRII and FZD-7 in HCC HepG2 and Huh-7 cells. The present
results demonstrated that simultaneously suppressing TGF-βRII and
FZD-7 significantly inhibited the proliferation of HepG2 and Huh-7
cells. To additionally investigate the possible mechanisms of
anti-proliferation efficacy, cell-cycle analysis was performed and
a high proportion of cells at G1 phase arrest were observed
following a blockade of TGF-βRII and FZD-7 in Huh-7 cells. The DNA
content of the cells reflects the specific processes of cell growth
and proliferation. In the cell cycle, cells go through various
stages of DNA replication: G0 phase, cells are in a quiescent
state; G1 phase, cells undergo pre-DNA synthesis; S phase, DNA is
synthesized by the cells; G2 phase, DNA in the cells becomes
tetraploid and cells reserve energy for mitosis (22). There are various checkpoints for the
cells during the cell cycle. One checkpoint is the G1/S transition
checkpoint, following which cells are no longer dependent on
exogenous proliferative stimulation; therefore; the cells acquire
the capacity to independently complete the cell cycle (22). Cyclin D1 forms a complex with
cyclin-dependent kinases 4 and 6 and functions as a regulatory
subunit of this complex. Cyclin D1 is required to facilitate the
transition between G1 and S phases; therefore, it promotes the
proliferation of cells and may contribute to tumorigenesis
(23,24). In addition, cyclin D1 acts as a
downstream effector molecule for Wnt/β-catenin signaling. In the
present study, the decrease in cyclin D1 expression following RNA
interference contributed to the cells arresting at the G1 phase,
which may have prevented the proliferation of HepG2 and Huh-7
cells. As another direct target gene of the Wnt/β-catenin signaling
pathway, c-Myc is also an oncogenic transcription factor, which
participates in a broad spectrum of physiological and pathological
processes, including cell proliferation, apoptosis,
differentiation, senescence and angiogenesis (25). c-Myc regulates cells in the G1 phase
to progress by positively regulating the activity of cyclin
dependent kinases (26–28). Consequently, a suppression of c-Myc
expression may reduce the proliferation and genesis of tumors. The
present results revealed that the downregulation of cyclin D1 and
c-Myc led to cells arresting at the G1 phase; therefore,
contributing to the inhibition of the proliferative abilities of
the cells, which is consistent with previous studies.
Cell invasion and migration are involved in a number
of physiological processes. However, uncontrolled cell migration
and invasion may lead to the development of metastasis, which is
the cause of ~90% of human cancer-associated mortalities (29). Invasion and migration of cancer cells
may be facilitated by TGF-β through various mechanisms, including
regulating cell survival, angiogenesis, vascular integrity and
interacting with the tumor microenvironment (21). An additional mechanism hypothesized to
contribute to metastasis is reversible EMT, through which
epithelial cells differentiate into mesenchymal cells (30). EMT usually occurs in tissue
morphogenesis during cell development, wound repair and cancer
progression in adult tissues (31).
TGF-β has been demonstrated to induce a morphological alteration,
which affects EMT. The transdifferentiation is accompanied by a
reorganization of the actin cytoskeleton, downregulation of
adhesion and cytoskeleton molecules and other extracellular matrix
associations in the cells, which eventually leads to enhanced
migratory and invasive properties of the cell (16). TGF-β may also contribute to the
tyrosine phosphorylation of β-catenin, which disrupts the
E-cadherin-β-catenin complex leading to the promotion of migration
and invasive abilities of the cell (32). In the present study, silencing of
TGF-β and Wnt/β-catenin signaling may lead to the
E-cadherin-β-catenin complex stimulating the invasion and migration
in HepG2 and Huh-7 cells.
Overall, the present results suggest that
simultaneous targeting of TGF-β and Wnt/β-catenin signaling
pathways exerts synergistic anti-tumor effects on HCC cells.
Additional in vitro, in vivo and clinical studies are
required to reveal more information concerning these pathways in
the tumorigenesis of cells. The present findings may support the
development of a novel treatment strategy that has greater efficacy
in the management of HCC.
Acknowledgements
The present study was supported by the Health
Department Foundation of Jiangsu Province (grant no. H201322) and
the Program for the Six Talents Peak in Jiangsu Province (grant no.
WS-080).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eskens FA, van Erpecum KJ, de Jong KP, van
Delden OM, Klumpen HJ, Verhoef C, Jansen PL, van den Bosch MA,
Méndez Romero A, Verheij J, et al: Hepatocellular carcinoma: Dutch
guideline for surveillance, diagnosis and therapy. Neth J Med.
72:299–304. 2014.PubMed/NCBI
|
|
3
|
Lee HS, Park MH, Yang SJ, Park KC, Kim NS,
Kim YS, Kim DI, Yoo HS, Choi EJ and Yeom YI: Novel candidate
targets of Wnt/beta-catenin signaling in hepatoma cells. Life Sci.
80:690–698. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ogasawara N, Tsukamoto T, Mizoshita T,
Inada K, Cao X, Takenaka Y, Joh T and Tatematsu M: Mutations and
nuclear accumulation of beta-catenin correlate with intestinal
phenotypic expression in human gastric cancer. Histopathology.
49:612–621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ozaki S, Ikeda S, Ishizaki Y, Kurihara T,
Tokumoto N, Iseki M, Arihiro K, Kataoka T, Okajima M and Asahara T:
Alterations and correlations of the components in the Wnt signaling
pathway and its target genes in breast cancer. Oncol Rep.
14:1437–1443. 2005.PubMed/NCBI
|
|
6
|
Mårtensson A, Öberg A, Jung A, Cederquist
K, Stenling R and Palmqvist R: Beta-catenin expression in relation
to genetic instability and prognosis in colorectal cancer. Oncol
Rep. 17:447–452. 2007.PubMed/NCBI
|
|
7
|
Fuchs SY, Ougolkov AV, Spiegelman VS and
Minamoto T: Oncogenic beta-catenin signaling networks in colorectal
cancer. Cell Cycle. 4:1522–1539. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Willert K and Jones KA: Wnt signaling: Is
the party in the nucleus? Genes Dev. 20:1394–1404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Holland JD, Klaus A, Garratt AN and
Birchmeier W: Wnt signaling in stem and cancer stem cells. Curr
Opin Cell Biol. 25:254–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ye N, Wang B, Quan ZF, Pan HB, Zhang ML
and Yan QG: The research progress of the interactions between miRNA
and Wnt/beta-catenin signaling pathway in breast cancer of human
and mice. Asian Pac J Cancer Prev. 15:1075–1079. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roberts AB and Wakefield LM: The two faces
of transforming growth factor beta in carcinogenesis. Proc Natl
Acad Sci USA. 100:8621–8623. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan Y, Xu Q, Li Y, Mao X and Zhang K:
Crosstalk between the p38 and TGF-β signaling pathways through
TβRI, TβRII and Smad3 expression in plancental choriocarcinoma
JEG-3 cells. Oncol Lett. 8:1307–1311. 2014.PubMed/NCBI
|
|
13
|
Miyazono K, Suzuki H and Imamura T:
Regulation of TGF-beta signaling and its roles in progression of
tumors. Cancer Sci. 94:230–234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Labbé E, Letamendia A and Attisano L:
Association of Smads with lymphoid enhancer binding factor 1/T
cell-specific factor mediates cooperative signaling by the
transforming growth factor-beta and wnt pathways. Proc Natl Acad
Sci USA. 97:8358–8363. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Z, Zhang LJ, Zhang HR, Tian GF, Tian J,
Mao XL, Jia ZH, Meng ZY, Zhao LQ, Yin ZN and Wu ZZ: Tumor-derived
transforming growth factor-β is critical for tumor progression and
evasion from immune surveillance. Asian Pac J Cancer Prev.
15:5181–5186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nishita M, Hashimoto MK, Ogata S, Laurent
MN, Ueno N, Shibuya H and Cho KW: Interaction between Wnt and
TGF-beta signalling pathways during formation of Spemann's
organizer. Nature. 403:781–785. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Labbé E, Lock L, Letamendia A, Gorska AE,
Gryfe R, Gallinger S, Moses HL and Attisano L: Transcriptional
cooperation between the transforming growth factor-β and Wnt
pathways in mammary and intestinal tumorigenesis. Cancer Res.
67:75–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer - a double-edged sword. Trends Cell Biol.
11:S44–S51. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Howard S, Deroo T, Fujita Y and Itasaki N:
A positive role of cadherin in Wnt/β-catenin signalling during
epithelial-mesenchymal transition. PLoS One. 6:e238992011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Merle P, de la Monte S, Kim M, Herrmann M,
Tanaka S, Von Dem Bussche A, Kew MC, Trepo C and Wands JR:
Functional consequences of frizzled-7 receptor overexpression in
human hepatocellular carcinoma. Gastroenterology. 127:1110–1122.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miyazono K, Ehata S and Koinuma D:
Tumor-promoting functions of transforming growth factor-β in
progression of cancer. Ups J Med Sci. 117:143–152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh AM and Dalton S: The cell cycle and
Myc intersect with mechanisms that regulate pluripotency and
reprogramming. Cell Stem Cell. 5:141–149. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ferraz C, Lorenz S, Wojtas B, Bornstein
SR, Paschke R and Eszlinger M: Inverse correlation of miRNA and
cell cycle-associated genes suggests influence of miRNA on benign
thyroid nodule tumorigenesis. J Clin Endocrinol Metab. 98:E8–E16.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi QQ, Zuo GW, Feng ZQ, Zhao LC, Luo L,
You ZM, Li DY, Xia J, Li J and Chen DL: Effect of trichostatin A on
anti HepG2 liver carcinoma cells: Inhibition of HDAC activity and
activation of Wnt/β-Catenin signaling. Asian Pac J Cancer Prev.
15:7849–7855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lan FF, Wang H, Chen YC, Chan CY, Ng SS,
Li K, Xie D, He ML, Lin MC and Kung HF: Hsa-let-7g inhibits
proliferation of hepatocellular carcinoma cells by downregulation
of c-Myc and upregulation of p16(INK4A). Int J Cancer. 128:319–331.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liao DJ, Thakur A, Wu J, Biliran H and
Sarkar FH: Perspectives on c-Myc, Cyclin D1, and their interaction
in cancer formation, progression, and response to chemotherapy.
Crit Rev Oncog. 13:93–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shachaf CM, Kopelman AM, Arvanitis C,
Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B,
Cardiff RD, et al: MYC inactivation uncovers pluripotent
differentiation and tumour dormancy in hepatocellular cancer.
Nature. 431:1112–1117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Giuriato S, Ryeom S, Fan AC, Bachireddy P,
Lynch RC, Rioth MJ, van Riggelen J, Kopelman AM, Passegué E, Tang
F, et al: Sustained regression of tumors upon MYC inactivation
requires p53 or thrombospondin-1 to reverse the angiogenic switch.
Proc Natl Acad Sci USA. 103:16266–16271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brabletz T, Hlubek F, Spaderna S,
Schmalhofer O, Hiendlmeyer E, Jung A and Kirchner T: Invasion and
metastasis in colorectal cancer: Epithelial-mesenchymal transition,
mesenchymal-epithelial transition, stem cells and β-catenin. Cells
Tissues Organs. 179:56–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim H, Yoo SB, Sun P, Jin Y, Jheon S, Lee
CT and Chung JH: Alteration of the E-Cadherin/β-Catenin complex is
an independent poor prognostic factor in lung adenocarcinoma.
Korean J Pathol. 47:44–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|