Introduction
Pancreatic cancer is the fourth most common cause of
cancer-associated mortality worldwide (1). The disease is highly aggressive as it is
often discovered late and typically develops resistance to
conventional therapy (2). The
five-year survival rate is <5%, indicating a poor prognosis for
all stages (1,3).
In colorectal cancer and early breast cancer, it has
been found that the proportion of stromal cells within the primary
tumor has prognostic value (4,5).
Pancreatic cancer tissue is characterized by its enrichment of
stromal cells. Furthermore, pancreatic stellate cells, which are
myofibroblast-like cells found in the areas of the pancreas that
have exocrine function, are critical in the desmoplastic reaction,
resulting in cancer cell progression (6). However, it has not been investigated
whether the pancreatic cancer cell-to-stromal cell ratio in the
primary tumor is a prognostic factor.
In cancer biology, the nature of the cancer
microenvironment is important (7).
The cancer microenvironment, which is formed by tumor cells and
tumor-mediated interactions with stromal cells and the
extracellular matrix (7), supports
malignant growth and invasion (6).
Stromal fibroblasts promote pancreatic cancer cell proliferation
(8). In squamous cell carcinoma,
fibroblasts lead collective cancer cell invasion into
three-dimensional (3D) culture (9).
For the analysis of cellular function and interaction, 3D culture
systems have been developed to mimic the in vivo
architecture of the cancer microenvironment in natural organs and
tissues (10).
Tumor budding is defined as the presence of
individual or small clusters (1–5 cells) of de-differentiated
cancer cells around the invasive front (11,12).
Budding is an independent prognostic factor in pancreatic cancer,
as well as colorectal and esophageal cancers (11,13,14). Tumor
budding is closely correlated with nodal metastasis (15), and is thought to reflect the process
of epithelial-mesenchymal transition (EMT), which increases the
capacity for migration and invasion (16,17).
Mammalian target of rapamycin (mTOR) controls the
size of cells through its modulation of the rate at which ribosomal
proteins are synthesized (18). mTOR
expression is associated with cancer progression and
chemoresistance (19). Therefore,
investigating the expression of mTOR in pancreatic cancer is
important in order to better understand the biology of the
disease.
In the present study, a 3D co-culture that mimicked
the microenvironment of pancreatic cancer was established. The
co-culture consisted of human pancreatic cancer cells (BxPC-3 cell
line) and skin fibroblasts (ASF-4-1 cell line) with extracellular
matrix collagen gel and Matrigel. The effect of a fibroblast-rich
environment on the malignant potential of pancreatic cancer was
investigated by analyzing tumor budding and mTOR expression through
immunohistochemical staining of the culture.
Materials and methods
Cells and cell culture
The human pancreatic cancer cell line BxPC-3 was
obtained from the RIKEN Bioresource Center Cell Bank (Tsukuba,
Japan), and human skin fibroblast ASF-4-1 cells were obtained from
the Japanese Collection of Research Bioresources Cell Bank
(National Institutes of Biomedical Innovation, Health and
Nutrition; Osaka, Japan).
Cells were cultured in RPMI-1640 medium
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal
bovine serum (FBS; Invitrogen, Thermo Fisher Scientific, Carlsbad,
CA, USA) and antibiotics (100 U/ml penicillin and 100 µg/ml
streptomycin; Nacalai Tesque, Inc., Kyoto, Japan) in a humidified
atmosphere of 5% CO2 at 37°C.
3D Matrigel and collagen invasion
assay
Matrigel (#354234; BD Biosciences, Franklin Lakes,
NJ, USA) was diluted at a 1:3 ratio with AteloCell (#KOU-RPM-02;
Koken Co., Ltd., Tokyo, Japan) and mixed. The mixture was then
placed in Falcon Cell Culture Inserts (8 µl pore size; #353097;
Corning, Inc., Corning, NY, USA) in 24-well plates (100 µl/well).
Following incubation for 1 h at 37°C, 500 µl of supplemented
RMPI-1640 medium (described above) was added to the bottom of each
well. BxPC-3 cells were adjusted to a final concentration of
1.0×106 cells/ml with FBS-free medium, and suspended
gently on the 3D Matrigel (as described above) for the collagen
invasion assay. Each well contained 1.0×105 pancreatic
cancer cells. For the addition of ASF-4-1 cells, the number of
these fibroblasts was adjusted to a 1:1 (fibroblast-poor) or 3:1
(fibroblast-rich) ratio with BxPC-3 cells in 100 µl of medium.
Gemcitabine (10 µM) was added 24 h after seeding, by which point
all cells had aggregated.
Following a 48-h incubation, the cells were assessed
under light microscopy (BX50F; Olympus Corporation, Tokyo, Japan).
The polycarbonate membranes at the bottom of each chamber were cut
and fixed in 10% formalin for 6 h, and subsequently fixed with HOLD
GEL 110 (Ebis1 kit; Asiakizai Co., Ltd., Tokyo, Japan) and embedded
in paraffin. Blocks were sliced into 3 µm-thick sections, stained
with hematoxylin and eosin and subjected to immunohistochemistry.
Invasion assays were performed a minimum of three times.
Immunohistochemistry and reagents
Automated immunohistochemical staining was performed
with a BenchMark LT slide stainer (Ventana Medical Systems, Inc.,
Tucson, AZ, USA). After pretreatment with citrate buffer (Ventana
Medical Systems, Inc.) for 60 min, sections were incubated for 32
min at 37°C with the following primary antibodies: Mouse monoclonal
antibodies against cytokeratin 18 (CK18; #sc-6259; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; dilution, 1:100), E-cadherin
(#18-0223; Invitrogen, Thermo Fisher Scientific; dilution, 1:50),
caspase-cleaved CK18 (M30 CytoDEATH; #10700; PEVIVA, Stockholm,
Sweden; dilution, 1:100), vimentin (#M0725; Dako, Glostrup,
Denmark; dilution, 1:100), Ki-67 (MIB-1; #08-1156; Invitrogen,
Thermo Fisher Scientific; ready-to-use); rabbit polyclonal
anti-CD31 (#ab28364; Abcam, Cambridge, MA, USA; dilution, 1:40) and
rabbit monoclonal anti-mTOR (#2983; Cell Signaling Technology,
Inc., Danvers, MA, USA; dilution, 1:100). The mTOR inhibitor
rapamycin was obtained from Abcam (#ab120224). Gemcitabine was
purchased from Wako (#077-05671; Wako Pure Chemical Industries,
Ltd., Osaka, Japan). Ki-67 proliferation indices were measured by
nuclear staining with the MIB-1 monoclonal antibody. For
chromogenic detection iVIEW DAB Detection kit (#760-091; Ventana
Medical Systems, Inc.) was used. This kit included secondary
antibodies, which were a mixture of biotin-conjugated goat
anti-mouse immunoglobulin (Ig)G polyclonal antibody, anti-mouse IgM
polyclonal antibody and anti-rabbit IgG polyclonal antibody.
Samples were incubated with secondary antibody at 37°C for 8 min.
The chromogenic reaction was performed using 3,3′-diaminobenzidine
and horseradish peroxidase-conjugated streptavidin (both taken from
the iVIEW DAB Detection kit). Evaluation of mTOR expression at the
invasive front was scored by the percentage and intensity of
staining. Staining percentage was scored as follows: 0 points,
<1%; 1 point, 1–20%; 2 points, 21–50%; and 3 points, >50%.
Staining intensity was scored as follows: 3 points, strong; 2
points, intermediate; 1 point, weak; and 0 points, negative. The
sum of these two scores served as the total score.
Xenograft and tissue preparation
Male severe combined immunodeficiency (SCID) mice
(n=20; weight, 23–25 g) were housed (5 mice per cage) in
pathogen-free conditions and exposed to 12 h light/dark cycles,
with ad libitum access to food and water. At 8 weeks of age
the mice were used. For implantation, an incision was made in the
backs of the mice, and 1.0×105 BxPC-3 cells with ASF-4-1
cells (1:1 or 1:3 ratio) from the 48-h 3D co-cultures were
transplanted into the subcutaneous tissue of the mice. Every week,
tumor sizes were recorded. After 4, 6, 8 or 10 weeks, five mice
were sacrificed by CO2 inhalation, the tumors were
harvested, embedded in paraffin and cut into 3-µm sections. The
sections were then stained with hematoxylin and eosin for
immunohistochemical analysis.
All animal experiments were performed in accordance
with the guidelines approved by the ethics committees of Mie
University (Tsu, Japan).
Statistical analysis
Mean tumor size and area were compared between
groups, and a two-tailed independent sample Student's t-test was
applied. P<0.01 was considered to indicate a statistically
significant difference.
Results
Fibroblast-rich co-cultures exhibit
tumor budding and greater resistance of BxPC-3 cells to gemcitabine
than cells in fibroblast-poor co-cultures
To analyze the association between pancreatic cancer
cells and fibroblasts, a 3D co-culture system closely mimicking the
physiological interactions in tissue was established. BxPC-3 cells
co-cultured with an equal number of ASF-4-1 cells were able to
migrate into the gel, and formed a characteristic invasive front at
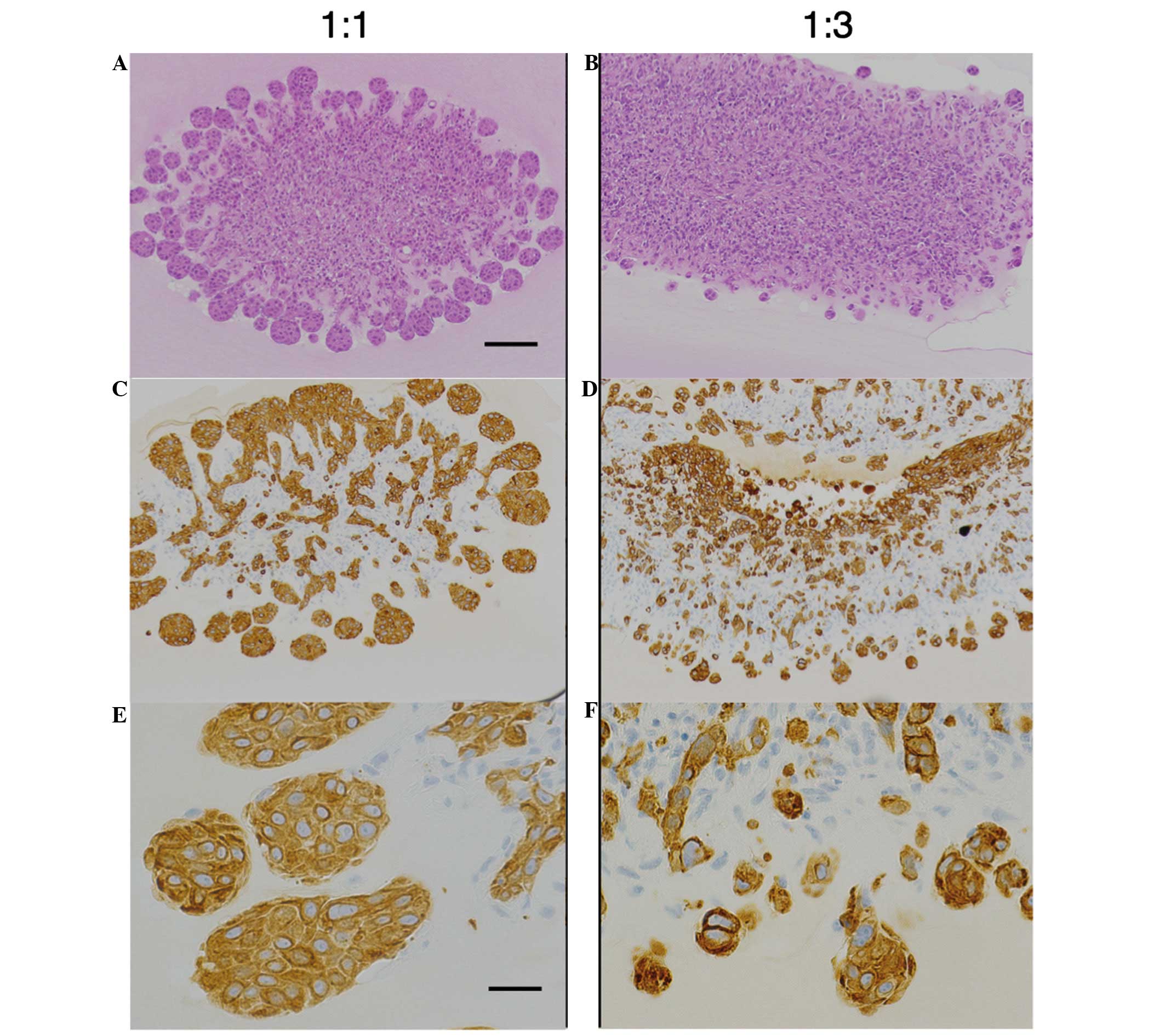
the advancing edge of the tumor (Fig.
1A–C). The invasive front was defined as the most external
cancer cell nest. To study whether fibroblasts promoted the
malignant potential of BxPC-3 cells (‘B’), increased numbers of
ASF-4-1 cells (‘A’) were co-cultured with a constant number of
BxPC-3 cells. In B:A co-cultures with a 1:3 ratio, tumor budding
was observed around the invasive front (Fig. 1D–F). Budding was defined as small
clusters of de-differentiated cancer cells around the invasive
front of the lesion; this correlates with poor prognosis in
pancreatic cancer as well as colorectal and esophageal cancers
(12–14). The number of buds was higher in
B:A=1:3 co-cultures than in B:A=1:1 co-cultures, as demonstrated by
CK18-positive findings, which indicate the distribution of cancer
cells (Fig. 1E and F).
Gemcitabine (10 µM) was added 24 h after seeding, by
which point all cells had aggregated. Following a further 24 h,
apoptotic/CK18-positive cells were counted. As observed in the
first part of the experiment, almost all cells at the invasive
front were cancer cells (Fig. 1C and
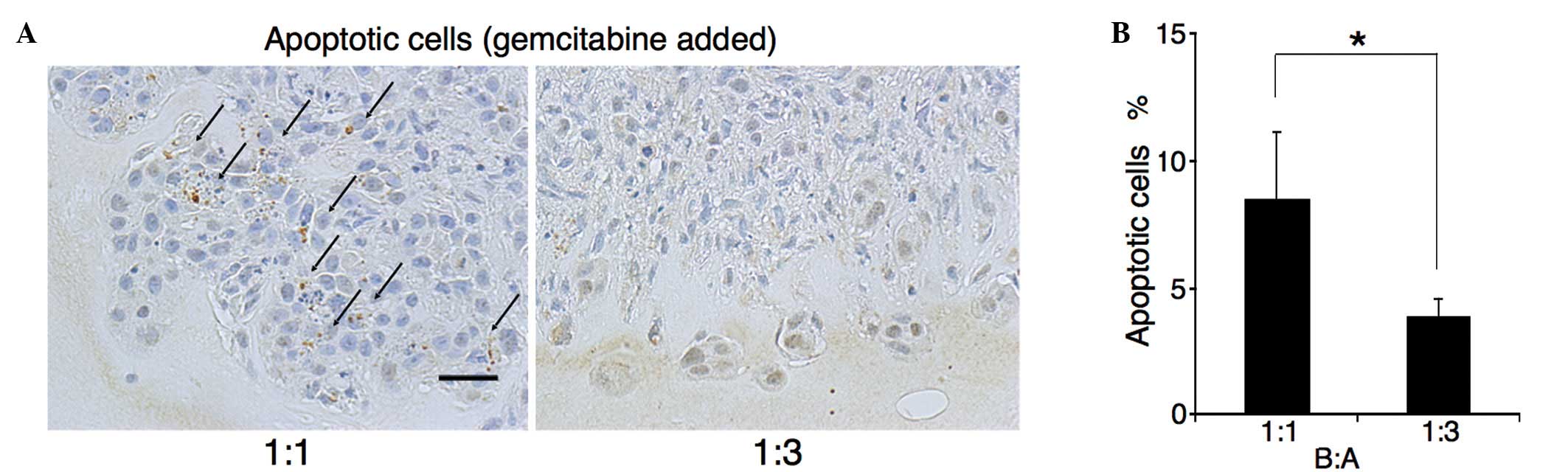
F). The M30 CytoDEATH antibody recognizes human caspase-cleaved
CK18, which is not observed in viable cells (20). The percentage of M30
CytoDEATH-positive cells at the invasive front was assessed
(Fig. 2A). In B:A=1:1 co-cultures,
the percentage of M30 CytoDEATH-positive cells was 8.5%, whilst
that in B:A=1:3 co-cultures was 3.9% (P=0.003) (Fig. 2B). Thus, BxPC-3 cells in B:A=1:3
co-cultures were more resistant to gemcitabine than cells in
B:A=1:1 co-cultures.
BxPC-3 cells at the invasive front of
fibroblast-rich co-cultures are larger than that in fibroblast-poor
cultures
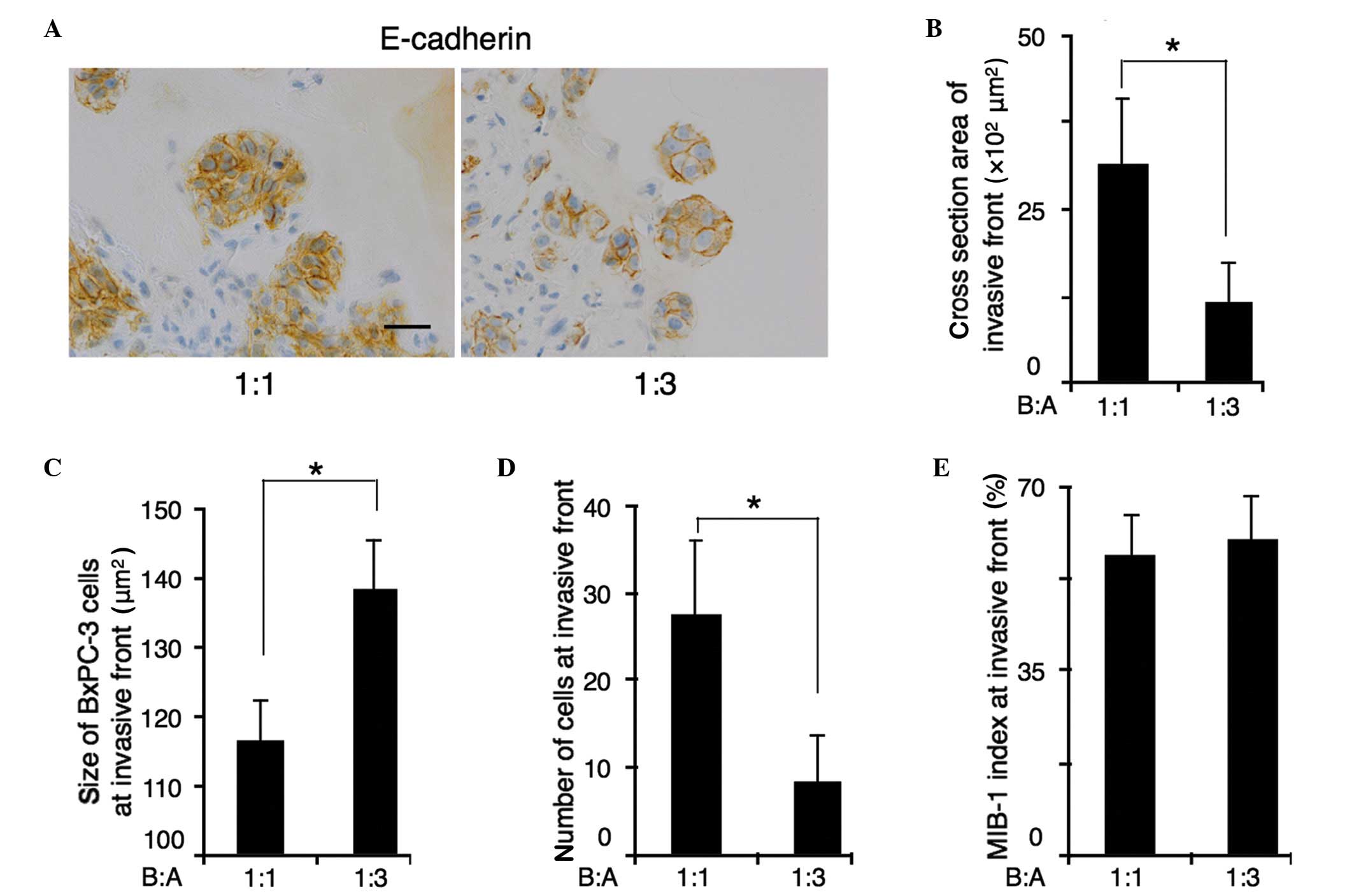
Immunohistochemical staining for E-cadherin was used
to allow measurement of the cross sectional area of the invasive
front. The cross sectional area was calculated using the image
analyzing software, DP2-BSW (Olympus Corporation), by tracing the
outline of invasive front. The size of BxPc-3 cells was calculated
by dividing the cross sectional area of the invasive front by the
number of BxPc-3 cells at the invasive front (Fig. 3A). The cross-sectional area of the
invasive front was decreased in B:A=1:3 co-cultures compared with
B:A=1:1 co-cultures. The average area of the cross-section of the
invasive front was 3,156 µm2 in B:A=1:1 co-cultures, and
1,155 µm2 in B:A=1:3 co-cultures (P<0.001) (Fig. 3B). Furthermore, the number of the
cells at the invasive front was decreased in B:A=1:3 co-cultures.
In B:A=1:1 co-cultures, the average number of BxPC-3 cells at the
invasive front was 23, whereas in B:A=1:3 co-cultures showed an
average of 8 cells (P<0.001) (Fig.
3C). Reducing the number of cells in the invasive front was
responsible for decreasing the size of the invasive front in
B:A=1:3 co-cultures. However, the average size of each BxPC-3 cell
at the invasive front of B:A=1:3 co-cultures was 138±5.8
µm2, which were larger than that of B:A=1:1 co-cultures
(116±5.8 µm2), despite the reduced cross-sectional area
of the invasive front (Fig. 3D).
MIB-1 indices remained unaltered, regardless of the number of
fibroblasts (Fig. 3E).
mTOR is increased at the invasive
front in fibroblast-rich co-cultures, and mTOR inhibition
suppresses invasive front formation and proliferation
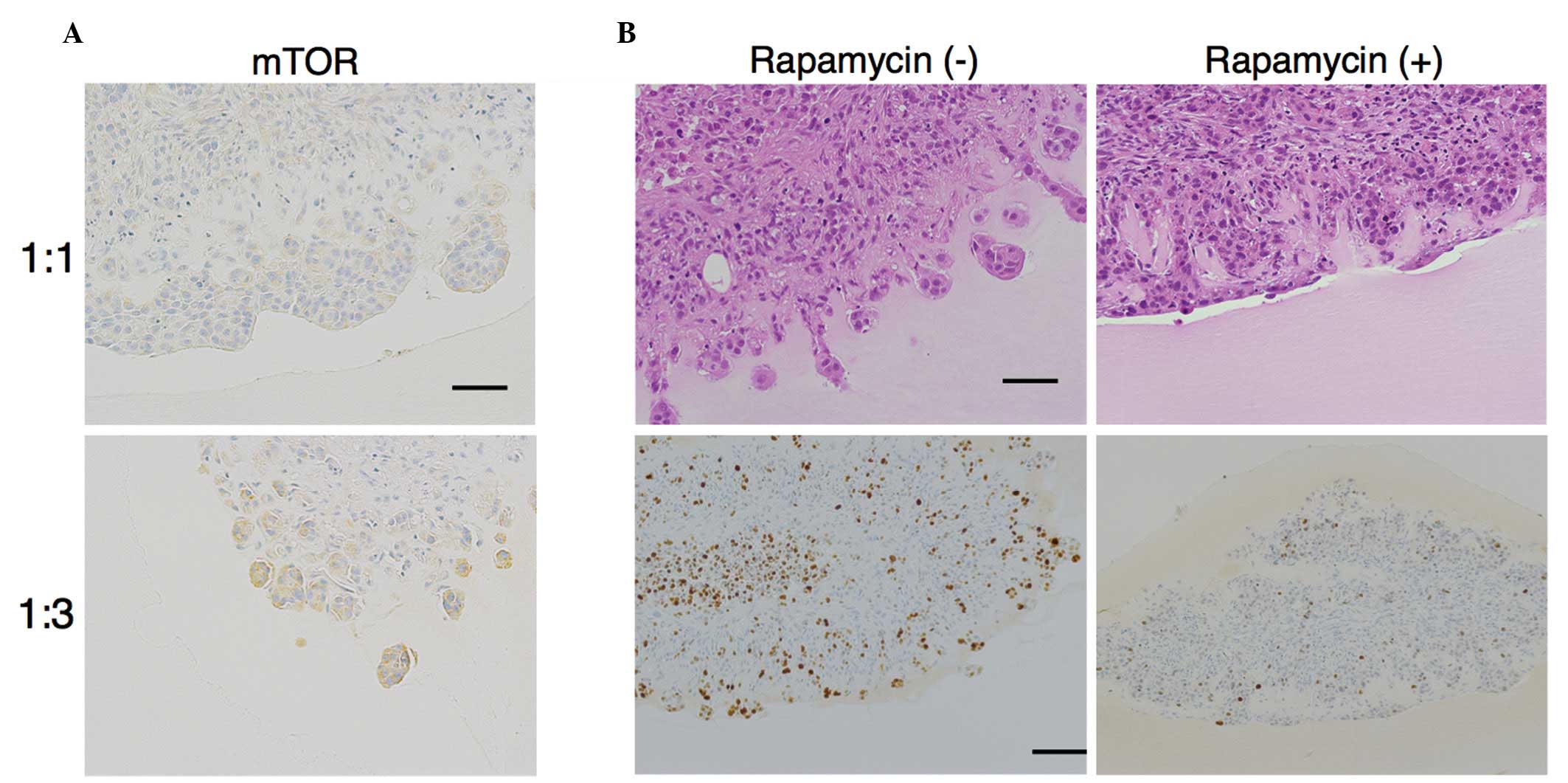
As mTOR controls the size of cells through
regulating the rate of protein synthesis of ribosomes, we
hypothesized that the increased size of cells at the invasive front
in B:A=1:3 co-cultures was related to mTOR. Immunohistochemical
staining revealed that expression of mTOR was increased at the
invasive front in B:A=1:3 co-cultures (total score = 5.666±0.516)
relative to that in B:A=1:1 co-cultures (total score = 0.666±1.032)
(Fig. 4A).
Treatment with rapamycin (100 nM), an mTOR
inhibitor, was observed to prevent invasive front formation in
B:A=1:3 co-cultures (Fig. 4B). The
MIB-1 index at the edge of the front without rapamycin in
fibroblast-rich co-cultures was 50%, whereas the MIB-1 index with
rapamycin was 7% (Fig. 4B). Thus, the
proliferation rate differed significantly in the presence or
absence of rapamycin (P<0.001). These results indicate that mTOR
expression in BxPC-3 cells may modulate the malignant potential of
the cancer cells.
A fibroblast-rich environment promotes
tumor growth and invasion in xenograft models
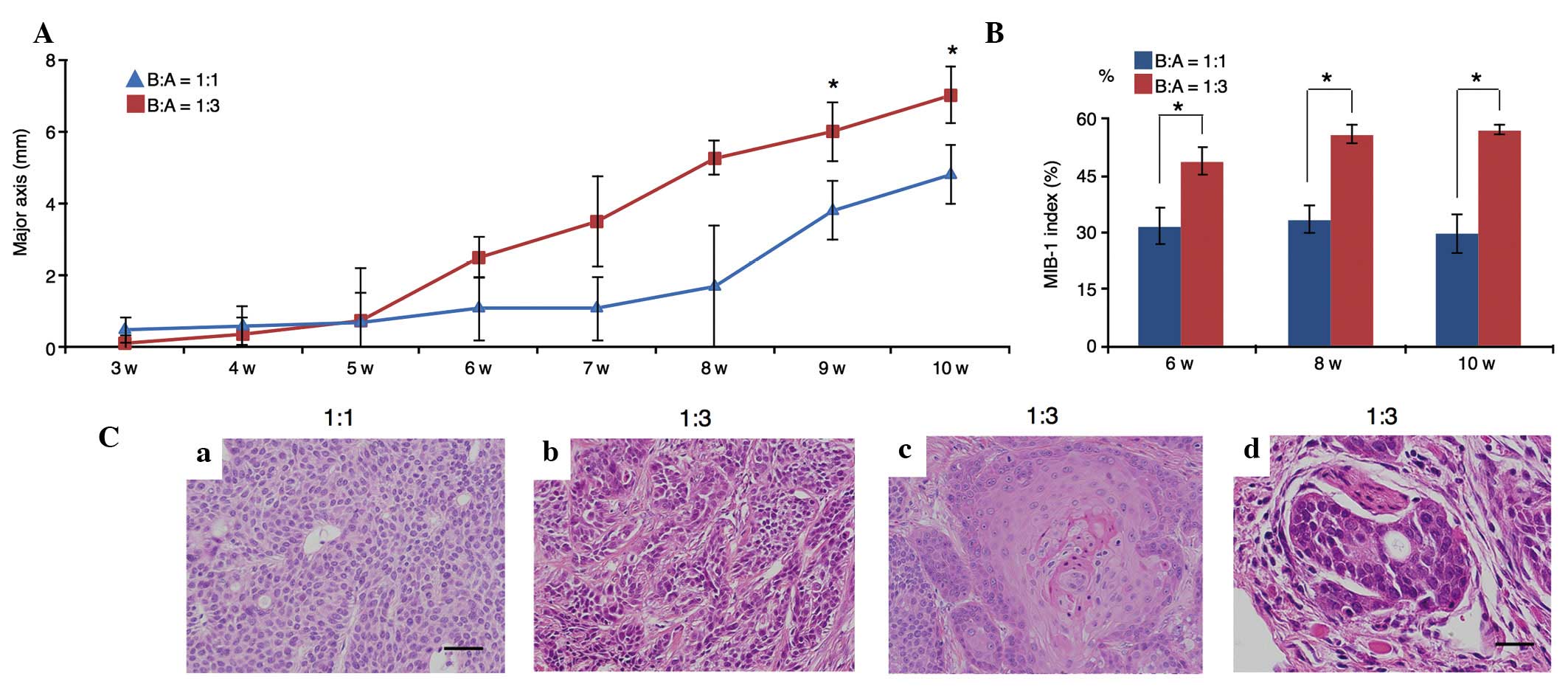
In xenograft models using SCID mice, tumor growth
initiated by B:A=1:3 co-cultures was more rapid than that initiated
by B:A=1:1 co-cultures (P=0.005; Fig.
5A). Additionally, the MIB-1 indices at 6, 8 and 10 weeks in
B:A=1:3 co-cultures were significantly higher than those in B:A=1:1
co-cultures (P=0.005; Fig. 5B).
Upon examination of the transplanted B:A=1:1
co-cultures, it was observed that BxPC-3 cells exhibited
homogeneous proliferation and had small, round nuclei; these cells
did not infiltrate into peripheral tissues (Fig. 5Ca). However, following transplantation
of B:A=1:3 co-cultures, BxPC-3 cells exhibited marked nuclear
atypia and pleomorphism (Fig. 5Cb).
Furthermore, differentiation to squamous cell carcinoma (Fig. 5Cc) and neural invasion (Fig. 5Cd) were observed in the B:A=1:3
co-culture group.
There was no significant difference in BxPC-3 mTOR
expression between B:A=1:1 and B:A=1:3 co-cultures in vivo
(data not shown). Angiogenesis was estimated by counting
CD31-positive vessels in the tumor; however, no significant
difference was observed (data not shown).
Discussion
The present study demonstrated that BxPC-3 cells
co-cultured with rich ASF-4-1 cells acquired chemoresistance,
invasive properties and increased mTOR expression in vitro.
Furthermore, fibroblast-rich culture promoted tumor growth and
invasion in vivo. This indicates that rich ASF-4-1 cells
promote the malignant potential of BxPC-3 cells in vitro and
in vivo.
In colon cancer and early breast cancer, the
enrichment of stromal cells within the primary tumor has been
observed to be correlated with poor prognosis (4,5). In
addition, various studies have concluded that the microenvironment
surrounding cancer cells promotes cancer progression (6,7).
Fibroblasts, in particular, support tumor growth and promote
metastasis and drug resistance (21,22).
Therefore, we hypothesized that a fibroblast-enriched
microenvironment would promote the malignant potential of the
pancreatic cancer cell line BxPC-3.
Tumor budding is an independent prognostic factor in
pancreatic cancer as well as other cancer types, particularly
colorectal cancer (12–14). Budding cells are defined as small
clusters that are de-differentiated and located around the invasive
front (11,12). These cells are CK18-positive and
suppress E-cadherin; therefore, they are thought to undergo EMT,
resulting in malignant progression (16). In the current study, budding cells
were observed around the invasive front in B:A=1:3 co-cultures.
When BxPC-3 cells migrated into the Matrigel-collagen mixture, they
appeared to have suppressed E-cadherin expression following their
interaction with fibroblasts, a process that may lead to tumor
budding. Under fibroblast-rich conditions, the number of BxPC-3
cells at the invasive front decreased compared with the B:A=1:1
co-cultures. Tumor budding is observed in pancreatic cancer in
addition to gastrointestinal carcinoma (11). In the present study, co-culture with a
high ratio of fibroblasts led BxPC-3 to initiate tumor budding and
possibly EMT.
The atypical serine/threonine protein kinase mTOR
belongs to the phosphoinositide-3 kinase (PI3K)-related kinase
family. mTOR forms two distinct complexes, named mTOR complex
(mTORC) 1 and 2 (18), by interacting
with a number of proteins. The mTORC1 pathway integrates at least
five major types of intracellular and extracellular signals (growth
factors, stress, energy status, oxygen and amino acid signals) to
control various major processes, including protein and lipid
synthesis, autophagy, proliferation, metabolism and cell growth
(18). In cancer cells, mTORC1
controls apoptosis and angiogenesis through hypoxia-inducible
factor 1 and vascular endothelial growth factor, and cell mobility
through the RhoA-Rac1 pathway (23,24). The
mTORC2 pathway integrates growth factors and controls metabolism,
cytoskeletal organization and cell survival (18). Thus, mTOR controls cell size in
addition to certain major signals in cancer cells, affecting
invasion, proliferation and migration. In the current study, the
mean size of the cells at the invasive front in B:A=1:3 co-cultures
was larger than that in B:A=1:1 co-cultures in vitro. In
addition, BxPC-3 cells at the invasive front expressed a higher
level of mTOR in B:A=1:3 co-cultures than did those in B:A=1:1
co-cultures in vitro. Furthermore, rapamycin, an mTOR
inhibitor, prevented BxPC-3 cells in B:A=1:3 co-cultures from
migrating and proliferating.
mTOR is involved in chemoresistance in pancreatic
cancer through an apoptotic pathway including nuclear factor κB and
the Akt/PI3K pathway (25,26). In the current study, an elevated
fibroblast ratio increased chemoresistance of BxPC-3 cells to
gemcitabine at the invasive front. BxPC-3 cells in the B:A=1:3
co-cultures may have suppressed apoptosis through the Akt/PI3K
pathway, resulting in increased chemoresistance. mTOR is downstream
from the Akt pathway. It has been shown that proliferation,
invasion and colony formation by BxPC-3 cells are promoted through
activation of mitogen-activated protein kinase (MAPK) and Akt
pathways by human pancreatic stellate cells (8). In the present study, it is suggested
that the Akt/PI3K pathway in BxPC-3 cells may have been activated
in the presence of high ratios of ASF-4-1 cells, resulting in
activation of mTOR. Consistently, in fibroblast-rich culture, mTOR
expression correlated with malignant potential of BxPC-3 cells.
In penile squamous cell carcinoma, mTOR has been
shown to be overexpressed in histologically high grade cases
(27). BxPC-3 cells with enriched
fibroblasts may have been in a histologically high grade due to
mTOR expression at the time of transplantation. In addition, neural
invasion of pancreatic cancer is correlated with phosphorylation of
the RET-Ras-MAPK pathway (28). As
mTOR is downstream from the Ras signal (18), in the current xenograft model, BxPC-3
cells from B:A=1:3 co-cultures may have had the ability for neural
invasion when they were transplanted. There was no significant
change in mTOR expression in B:A=1:1 co-cultures or B:A=1:3
co-cultures used for xenografting. It is possible that, following
transplantation, mTOR expression in BxPC-3 cells was induced by
some other factor, e.g., growth factors, cytokines, hormones, blood
flow or immunoregulation (18).
In the present study, the model tissue had no blood
flow and no immune response, which limited these results with
regard to the accuracy of mimicking of the pancreatic cancer
microenvironment in vivo. However, we suggest that this
model may be useful for cancer research, particularly studies of
the interaction between cancer cells and fibroblast enrichment, and
the mechanisms of tumor proliferation and invasion. Because the
passages of the cell lines were different, the ratio between cancer
cells and fibroblast cells may be altered due to their different
growth rate. However, the same experiments were repeated at least
three times with cells from different passages, yielding almost
identical Ki-67 indices; therefore, the difference of passages
exerts little influence on the result. In the future, we aim to
investigate the interactions with endothelial cells, immune cells,
other cell lines and pancreatic cancer cells derived from the human
body.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wolfgang CL, Herman JM, Laheru DA, Klein
AP, Erdek MA, Fishman EK and Hruban RH: Recent progress in
pancreatic cancer. CA Cancer J Clin. 63:318–348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Kruijf EM, van Nes JG, van de Velde CJ,
Putter H, Smit VT, Liefers GJ, Kuppen PJ, Tollenaar RA and Mesker
WE: Tumor-stroma ratio in the primary tumor is a prognostic factor
in early breast cancer patients, especially in triple-negative
carcinoma patients. Breast Cancer Res Treat. 125:687–696. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mesker WE, Junggeburt JM, Szuhai K, de
Heer P, Morreau H, Tanke HJ and Tollenaar RA: The carcinoma-stromal
ratio of colon carcinoma is an independent factor for survival
compared to lymph node status and tumor stage. Cell Oncol.
29:387–398. 2007.PubMed/NCBI
|
|
6
|
Dunér S, Lindman Lopatko J, Ansari D,
Gundewar C and Andersson R: Pancreatic cancer: The role of
pancreatic stellate cells in tumor progression. Pancreatology.
10:673–681. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liotta LA and Kohn EC: The
microenvironment of the tumour-host interface. Nature. 411:375–379.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hwang RF, Moore T, Arumugam T,
Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB and Logsdon CD:
Cancer-associated stromal fibroblasts promote pancreatic tumor
progression. Cancer Res. 68:918–926. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gaggioli C, Hooper S, Hidalgo-Carcedo C,
Grosse R, Marshall JF, Harrington K and Sahai E: Fibroblast-led
collective invasion of carcinoma cells with differing roles for
RhoGTPases in leading and following cells. Nat Cell Biol.
9:1392–1400. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin RZ and Chang HY: Recent advances in
three-dimensional multicellular spheroid culture for biomedical
research. Biotechnol J. 3:1172–1184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Karamitopoulou E, Zlobec I, Born D,
Kondi-Pafiti A, Lykoudis P, Mellou A, Gennatas K, Gloor B and Lugli
A: Tumour budding is a strong and independent prognostic factor in
pancreatic cancer. Eur J Cancer. 49:1032–1039. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ueno H, Price AB, Wilkinson KH, Jass JR,
Mochizuki H and Talbot IC: A new prognostic staging system for
rectal cancer. Ann Surg. 240:832–839. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Prall F: Tumour budding in colorectal
carcinoma. Histopathology. 50:151–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koike M, Kodera Y, Itoh Y, Nakayama G,
Fujiwara M, Hamajima N and Nakao A: Multivariate analysis of the
pathologic features of esophageal squamous cell cancer: Tumor
budding is a significant independent prognostic factor. Ann Surg
Oncol. 15:1977–1982. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Landau MS, Hastings SM, Foxwell TJ,
Luketich JD, Nason KS and Davison JM: Tumor budding is associated
with an increased risk of lymph node metastasis and poor prognosis
in superficial esophageal adenocarcinoma. Mod Pathol. 27:1578–1589.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zlobec I and Lugli A: Epithelial
mesenchymal transition and tumor budding in aggressive colorectal
cancer: Tumor budding as oncotarget. Oncotarget. 1:651–661. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Karamitopoulou E: Role of
epithelial-mesenchymal transition in pancreatic ductal
adenocarcinoma: Is tumor budding the missing link? Front Oncol.
3:2212013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arlt A, Müerköster SS and Schäfer H:
Targeting apoptosis pathways in pancreatic cancer. Cancer Lett.
332:346–358. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leers MP, Kölgen W, Björklund V, Bergman
T, Tribbick G, Persson B, Björklund P, Ramaekers FC, Björklund B,
Nap M, et al: Immunocytochemical detection and mapping of a
cytokeratin 18 neo-epitope exposed during early apoptosis. J
Pathol. 187:567–572. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tod J, Jenei V, Thomas G and Fine D:
Tumor-stromal interactions in pancreatic cancer. Pancreatology.
13:1–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feig C, Gopinathan A, Neesse A, Chan DS,
Cook N and Tuveson DA: The pancreas cancer microenvironment. Clin
Cancer Res. 18:4266–4276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen K, Ji L, Gong C, Ma Y, Yang L, Fan Y,
Hou M and Wang Z: Notoginsenoside Ft1 promotes angiogenesis via
HIF-1α mediated VEGF secretion and the regulation of PI3K/AKT and
Raf/MEK/ERK signaling pathways. Biochem Pharmacol. 84:784–792.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tan CY and Hagen T: Post-translational
regulation of mTOR complex 1 in hypoxia and reoxygenation. Cell
Signal. 25:1235–1244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arlt A, Gehrz A, Müerköster S, Vorndamm J,
Kruse ML, Fölsch UR and Schäfer H: Role of NF-kappaB and Akt/PI3K
in the resistance of pancreatic carcinoma cell lines against
gemcitabine-induced cell death. Oncogene. 22:3243–3251. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamacher R, Schmid RM, Saur D and
Schneider G: Apoptotic pathways in pancreatic ductal
adenocarcinoma. Mol Cancer. 7:642008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chaux A, Munari E, Cubilla AL, Hicks J,
Lecksell K, Burnett AL and Netto GJ: Immunohistochemical expression
of the mammalian target of rapamycin pathway in penile squamous
cell carcinomas: A tissue microarray study of 112 cases.
Histopathology. 64:863–871. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gil Z, Cavel O, Kelly K, Brader P, Rein A,
Gao SP, Carlson DL, Shah JP, Fong Y and Wong RJ: Paracrine
regulation of pancreatic cancer cell invasion by peripheral nerves.
J Natl Cancer Inst. 102:107–118. 2010. View Article : Google Scholar : PubMed/NCBI
|