Introduction
Adrenocortical carcinoma is a rare malignant tumor,
accounting for ~0.02% of global malignancies (1). Adrenocortical carcinoma has a bimodal
distribution by age, with one peak age before 5 years, and the
other between 40 and 50 years old, as well as a female predominance
(2). Sarcomatoid carcinomas, which
are comprised of sarcomatous and carcinomatous differentiation
components, are a notable kind of malignant tumor, commonly
occurring in the digestive (3) and
respiratory (4) tracts, and breasts
(5). This tumor is rarely observed in
the adrenal glands. Primary adrenal sarcomatoid carcinoma, an
extremely uncommon, aggressive malignant tumor, constitutes a
subgroup of adrenocortical carcinomas (6,7). The
typical symptoms associated with the disease are abdominal pain,
weight loss and lumbago, and surgery is the major modality for the
treatment of primary adrenal sarcomatoid carcinoma. Primary adrenal
sarcomatoid carcinoma is likely to exhibit distant metastasis and
patients often succumb to disease within 2 years following surgery
(8). The first report of primary
adrenal sarcomatoid carcinoma was in 1989 by Collina et al
(6). At the time of writing the
present study, to the best of our knowledge only 11 cases had
previously been reported (6,9–18). The
majority of those patients succumbed to disease within a year, due
to local recurrence or de novo metastases. Timely and
accurate diagnosis and effective treatments are therefore required.
The present study reports a case of primary sarcomatoid carcinoma
with lung metastasis, and reviews the previous literature in order
to identify the optimal management practices.
Case report
A 59-year-old man presented to the Department of
Urology, Peking University Shenzhen Hospital (Shenzhen, China) for
examination on April 28, 2014, with a 4-month history of asthenia
and weight loss, without fever, abdominal pain or other notable
medical history. The patient provided written informed consent for
the publication of their data. Upon initial physical examination,
the patient's blood pressure was 115/75 mmHg, however, he exhibited
no other symptoms that could be due to excessive levels of
steroidal hormones or catecholamine. Abdominal palpation revealed
no masses. Aldosterone (56.38 pg/ml; normal, 23.50–106.60 pg/ml)
and cortisol (8 AM, 13.96 µg/dl; normal, 7.20–18.20 µg/dl; 4 AM,
6.61 µg/dl; normal, 2.75–6.65 µg/dl; 12 AM, 0.96 µg/dl; normal,
0.00–1.54 µg/dl) levels were within the normal limits. Initial
blood tests revealed low hemoglobin levels (71 g/l; normal, 120–160
g/l). The serum levels of carbohydrate antigen 125 exceeded the
normal limits [211.10 units (U)/ml; normal, <35.00 U/ml], while
those of carbohydrate antigen 19–9 (2.05 U/ml; normal, <37.00
U/ml), carbohydrate antigen 15–3 (8.80 U/ml; normal, <31.30
U/ml), α-fetoprotein (1.15 ng/ml; normal, <13.40 ng/ml),
carcinoembryonic antigen (0.74 ng/ml; normal, 0.00–5.00 ng/ml) and
squamous cell carcinoma antigen (0.7 ng/ml; normal, 0.0–1.5 ng/ml)
were within the normal ranges. Abdominal ultrasonography (LOGIQ E9;
GE Healthcare, Little Chalfont, UK) revealed a right, capsuled
adrenal mass of 6.5×3.7 cm, with no obvious calcification or
liquefaction. A computed tomography (CT) scan, using a Sensation 16
multiple detector CT (Siemens Healthcare, Erlangen, Germany), of
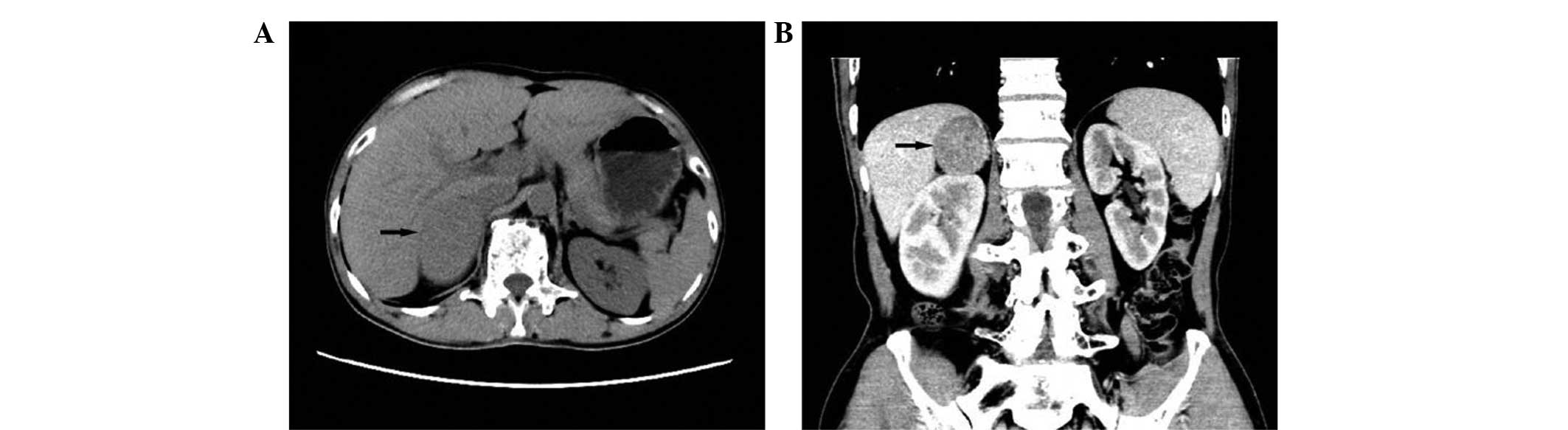
the abdomen indicated a well-defined, homogeneously dense, 6×4 cm
tumor in the right adrenal gland (Fig.
1). The tumor was adjacent to the liver, inferior vena cava and
right renal vein (Fig. 1). An
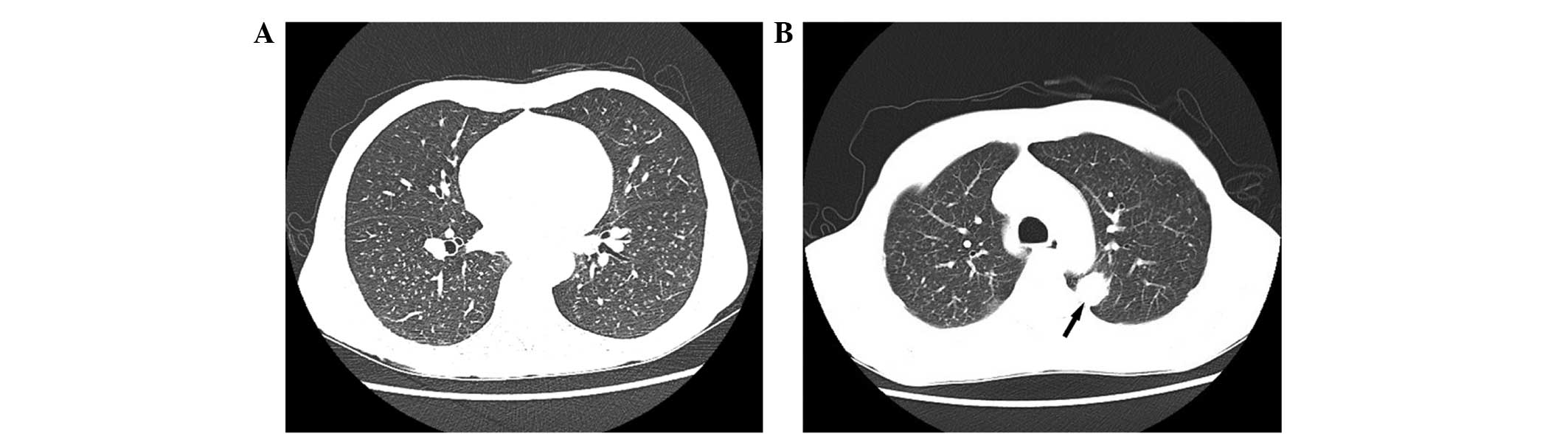
additional chest CT suggested bilateral lung metastases (Fig. 2). The preoperative diagnosis was
adrenal malignancy. The patient underwent right adrenalectomy. The
surgery revealed an entirely capsuled tumor adhering to the
liver.
Macroscopic examination revealed the tumor to be
soft and ~5 cm in diameter, with its dissected surface revealing a
white-to-grey rim and pink soft node within a well-defined capsule.
Following resection, tissues were sent to the Department of
Pathology, Peking University Shenzhen Hospital for pathological and
immunohistochemical analysis. Formalin (Guangzhou Kaixiu Co., Ltd,
Guangzhou, China)-fixed, paraffin (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China)-embedded resected tissues (4
µm pathological sections) were used for histological staining with
hematoxylin and eosin (Beijing Solarbio Science & Technology
Co., Ltd.). Tissues were visualized using a microscope (DMIL-LED;
Leica Microsystems, Wetzlar, Germany). Microscopic examination
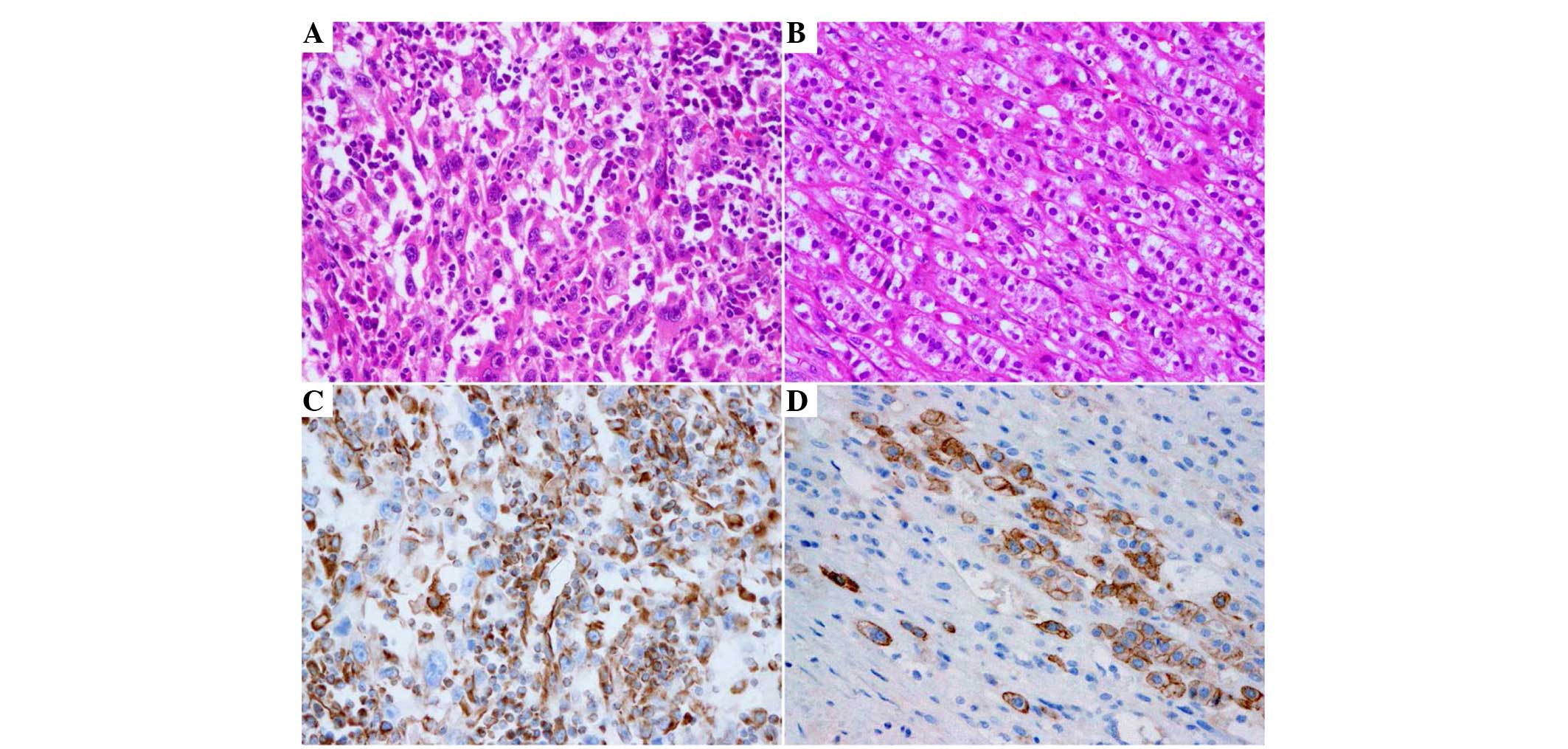
revealed extensive growth with epithelial and spindle cell
components (Fig. 3). Tumor cells were
observed to be large and stratified in sheets. Hyperchromatic
nuclei and atypical mitoses were present. Formalin-fixed,
paraffin-embedded tissues (4 µm pathological sections) were used
for immunohistochemical analysis with rabbit anti-vimentin
monoclonal (catalog no., RMA-0547; 1:500; Fuzhou Maixin Biotech.
Co., Ltd., Fuzhou, China) and mouse anti-cytokeratin monoclonal
(pan-Cytokeratin Immunohistochemistry Detection kit; catalog no.,
IPM067; 1:500; Guangzhou An Biping (LBP) Pharmaceutical Technology
Co., Ltd., Guangzhou, China) primary antibodies.
Immunohistochemistry revealed tumor cells that were diffused and
strongly positive for vimentin and cytokeratin (Fig. 3). Immunohistochemical stains for
smooth muscle actin, S-100 protein, α-inhibin, human melanoma black
45, melan-A protein, synaptophysin, chromogranin A, thyroid
transcription factor-1, desmin and myogenin were negative for
carcinomatous and sarcomatous components (Fig. 3). The final pathological diagnosis was
primary sarcomatoid carcinoma of the right adrenal gland.
Postoperatively, the patient refused adjuvant chemotherapy and was
discharged from hospital. At the time of writing, 6 months
postoperatively, the patient remained alive and, to the best of our
knowledge, was not undergoing any further treatment or follow-up
visits.
Discussion
Adrenocortical carcinoma is an uncommon, aggressive
tumor, occurring annually at the rate of 0.7–2.0/1,000,000 of the
global population (19), and is known
to metastasize through the lymphatic and circulatory systems, as
well as through implantation (20).
It has been reported that 21–39% of all patients have distant
metastasis at the time of diagnosis, which emphasizes the urgent
need for novel treatment methods (2,21).
Adrenocortical carcinoma typically metastasizes to the lung, liver,
peritoneum and pleura, lymph nodes, and bones (22). Prognosis is considerably poor, and the
5-year overall survival rate of advanced adrenocortical carcinoma
is <30% (23). Even with favorably
performed radical resections, the majority of patients succumb to
local recurrence or de novo metastases (2,8,21).
Adrenal sarcomatoid carcinoma is a rare
adrenocortical carcinoma subgroup (7). The 2004 WHO classification defines it as
a ‘sarcomatoid carcinoma’ (7). It
rarely occurs in the urinary system and morbidity has been observed
to be <3% for patients with urological malignancy (24). Sarcomatoid carcinomas are described as
malignant tumors comprised of epithelial and mesenchymal
components, which may malignantly differentiate into cartilage,
fibrosarcoma, bone or skeletal muscle (25–27).
Adrenal sarcomatoid carcinomas are more likely to occur in the
kidney or bladder, rather than the adrenal glands (17). These carcinomas may be classified as
either functional or nonfunctional tumors (1). Patients with functional tumors may
present with symptoms caused by excessive hormones, including the
following: i) Cushing's syndrome; ii) severe hypertension; iii)
male feminization or iv) female masculinization. Patients with
nonfunctional tumors may present with symptoms including the
following: i) Weight loss; ii) abdominal masses; iii) abdominal
pain; iv) low fever and v) asthenia (1).
To the best of our knowledge, the present case
report describes the 12th reported case of primary adrenal
sarcomatoid carcinoma and reviews the previous literature (Table I) (6,9–18). Data collected from the present
patient, as well as from the previous 11 reported cases identified
in the literature, revealed that the patient age range at the time
of presentation with adrenal sarcomatoid carcinoma was 29–79 years,
with a mean age of 55.75 years (6,9–18). The female:male ratio in this small
sample size was 1:1. The majority of studies have demonstrated that
women are more susceptible to the disease (1,28). Based
on the collected data, there appear to be no significant
differences in morbidity rates between male and female patients
(Table 1). In addition, based on the
collection data, middle-aged and elderly patients should be
considered high risk.
 | Table I.Reported cases of adrenal sarcomatoid
carcinomas from the literature. |
Table I.
Reported cases of adrenal sarcomatoid
carcinomas from the literature.
| First author | Patient age,
years | Gender | Symptoms | Endocrine
dysfunction | Size, cm | Sarcomatous
component | Metastasis at the
time of diagnosis | Postoperative
survival | Reference |
|---|
| Collina et
al | 68 | F | Abdominal
distention | No | 11 | Spindle cells | No | 6 months | (6) |
| Okazumi et
al | 46 | M | Abdominal
distention | No | 14 | Spindle cells | No | 6 months | (9) |
| Decorato et
al | 42 | F | Abdominal pain | No | 19 | Rhabdomyosarcoma | No | 7 months | (10) |
| Fischler et
al | 29 | F | Virilization,
Weight loss | Yes | 12 |
Rhabdomyosarcoma | No | 8 months | (11) |
| Barksdale et
al | 79 | F | Hypertension | Yes | 5 | Osteosarcoma,
Chondrosarcoma | Inferior vena
cava | 4 months | (12) |
| Lee et
al | 61 | M | Flank pain | No | 12 | Spindle cells | Liver | 2 days | (13) |
| Strum et
al | 31 | M | Abdominal pain | No | 12 | Spindle cells | No | 3 months | (14) |
| Sasaki et
al | 45 | M | Abdominal pain | No | 17 |
Rhabdomyosarcoma | No | 3 months | (15) |
| Coli et
al | 75 | F | Abdominal pain | No | 15 | Spindle cells | No | 12 months | (16) |
| Yan et
al | 72 | M | Flank pain, Low
fever | No | 13 | Spindle cells | No | 30 months | (17) |
| Shaikh et
al | 62 | F | Abdominal pain | No | 6 | Spindle cells | No | 3 months | (18) |
| Zhu et
al | 59 | M | Asthenia, Weight
loss | No | 6 | Spindle cells | Lung | Alive at time of
writing | Present case |
Upon initial presentation with primary adrenal
sarcomatoid carcinoma, patients typically complain of abdominal
pain or discomfort, lumbago and weight loss (6,9–18). Only 2/12 reported cases were
functional tumors (11,12). The hormone levels of the present
patient were within the normal limits. It is likely that, at the
time of diagnosis, the patients in all 12 cases were at an advanced
disease stage and had distant metastases. At diagnosis, 2 of the
reported cases presented with inferior vena cava invasion and liver
metastasis (12,13), while the present patient presented
with lung metastasis preoperatively. All 12 cases were surgically
treated. Only 3 of these cases received combined adjuvant
chemotherapy, including the present study (6,14). The
majority of patients succumbed to recurrence or metastasis
complications within a year of surgery. The patient in the present
study, along with 7 others from the literature (6,9,13,14,16–18),
demonstrated a sarcomatous component containing spindle cells,
while 4 had rhabdomyosarcoma (10–12,15), or a
mixture of osteosarcoma and chondrosarcoma.
Adrenal sarcomatoid carcinomas tend to be
aggressive, and patients often present with extensive locoregional
spread at diagnosis (1). Preoperative
diagnosis is challenging, which reduces treatment effectiveness and
directly influences prognosis. A previous study demonstrated that
endocrine assays and CT scans are effective modalities for
diagnosis and tumor location confirmation (29). Dynamic enhanced multi-detector row CT
scans may reveal tumor composition and blood supply (29). Histopathology and immunohistochemistry
must be performed to confirm a diagnosis (30). Surgical excision, including complete
metastatic resection, is currently the most effective treatment,
while postoperative adjuvant chemotherapy may decrease relapse and
metastasis rates (31). Previous case
reports (6,14) and the present study appear to suggest
that chemotherapy has no significant effect on adrenal sarcomatoid
carcinomas. Additional research is required in order to confirm the
efficacy of chemotherapy. Mitotane is recommended as the preferred
treatment for patients with advanced adrenocortical carcinoma
(32). It works primarily by inducing
adrenal cortex degeneration and necrosis. Chemotherapy combined
with mitotane has been demonstrated to be more effective than
chemotherapy alone (33). In these
treatment regimes, mitotane serum levels must be monitored.
Molecularly-targeted tumor therapy has previously been studied with
findings indicating that certain oncogenes, growth factors and
tumor-suppressor genes are associated with adrenocortical carcinoma
tumorigenesis. It should be noted that epidermal growth factor
receptor (EGFR) is overexpressed in the majority of adrenocortical
carcinomas (34), and has been
demonstrated to have a significant role in sarcomatoid carcinomas
(35,36). Anti-EGFR therapy may comprise a
feasible approach for the improvement of prognosis in patients with
adrenal sarcomatoid carcinomas.
Adrenal sarcomatoid carcinomas are rare but
aggressive tumors, and are associated with poor prognosis and a
lack of effective treatment options. At present, only postoperative
immunohistochemical examination is able to provide a definitive
diagnosis. Surgery is the single, yet ineffective, treatment option
for tumors at present. Adjuvant chemotherapy is ineffective for
advanced adrenal sarcomatoid carcinoma; therefore, additional
research is required in order to explore targeted therapy for the
treatment of this rare, but fatal, type of tumor.
Acknowledgements
The present study was supported by the Medical
Scientific Research Foundation of Guangdong Province, China (grant.
no. A2014653) and the Science and Technology project in Shenzhen,
Guangdong Province, China (grant. no. 201302052).
References
|
1
|
Roman S: Adrenocortical carcinoma. Curr
Opin Oncol. 18:36–42. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ng L and Libertino JM: Adrenocortical
carcinoma: Diagnosis, evaluation and treatment. J Urol. 169:5–11.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weidner N: Sarcomatoid carcinoma of the
upper aerodigestive tract. Semin Diagn Pathol. 4:157–168.
1987.PubMed/NCBI
|
|
4
|
Zarbo RJ, Crissman JD, Venkat H and Weiss
MA: Spindle-cell carcinoma of the upper aerodigestive tract mucosa.
An immunohistologic and ultrastructural study of 18 biphasic tumors
and comparison with seven monophasic spindle-cell tumors. Am J Surg
Pathol. 10:741–753. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oberman HA: Metaplastic carcinoma of the
breast. A clinicopathologic study of 29 patients. Am J Surg Pathol.
11:918–929. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Collina G, Maldarizzi F, Betts CM and
Eusebi V: Primary sarcomatoid carcinoma of the adrenal gland. First
case report. Virchows Arch A Pathol Anat Histopathol. 415:161–167.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
DeLellis RA, Lloyd RV, Heitz PU and Eng C:
World Health Organization Classification of Tumors. Pathology and
Genetics of Tumours of Endocrine Organs. 8:IARC Press. (Lyon,
France). 2004.
|
|
8
|
Tauchmanovà L, Colao A, Marzano LA,
Sparano L, Camera L, Rossi A, Palmieri G, Marzano E, Salvatore M,
Pettinato G, et al: Andrenocortical carcinomas: Twelve-year
prospective experience. World J Surg. 28:896–903. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okazumi S, Asano T, Ryu M, Nagashima T,
Odaka M, Isono K and Nishizawa T: Surgical resection of adrenal
carcinoma extending into the vena cava, right atrium and ventricle:
Case report and review of the literature. Nihon Geka Gakkai Zasshi.
88:231–238. 1987.(In Japanese). PubMed/NCBI
|
|
10
|
Decorato JW, Gruber H, Petti M and
Levowitz BS: Adrenal carcinosarcoma. J Surg Oncol. 45:134–136.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fischler DF, Nunez C, Levin HS, McMahon
JT, Sheeler LR and Adelstein DJ: Adrenal carcinosarcoma presenting
in a woman with clinical signs of virilization. A case report with
immunohistochemical and ultrastructural findings. Am J Surg Pathol.
16:626–631. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barksdale SK, Marincola FM and Jaffe G:
Carcinosarcoma of the adrenal cortex presenting with
mineralocorticoid excess. Am J Surg Pathol. 17:941–945. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee MS, Park IA, Chi JG, Ham EK, Lee KC
and Lee CW: Adrenal carcinosarcoma-a case report. J Korean Med Sci.
12:374–377. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sturm N, Moulai N, Laverrière MH, Chabre
O, Descotes JL and Brambilla E: Primary adrenocortical sarcomatoid
carcinoma: Case report and review of literature. Virchows Arch.
452:215–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sasaki K, Desimone M, Rao HR, Huang GJ and
Seethala RR: Adrenocortical carcinosarcoma: A case report and
review of the literature. Diagn Pathol. 5:512010.PubMed/NCBI
|
|
16
|
Coli A, Di Giorgio A, Castri F, Destito C,
Marin AW and Bigotti G: Sarcomatoid carcinoma of the adrenal gland:
A case report and review of literature. Pathol Res Pract.
206:59–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan JJ, Sun AJ, Ren Y and Hou C: Primary
adrenocortical sarcomatoid carcinoma: Report of a case. Can Urol
Assoc J. 6:E189–E191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shaikh AS, Bakhshi GD, Khan AS, Jamadar
NM, Nirmala AK and Raza AA: Primary adrenal sarcomatoid carcinoma.
Clin Pract. 4:6042014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fassnacht M, Libé R, Kroiss M and Allolio
B: Adrenocortical carcinoma: A clinician's update. Nat Rev
Endocrinol. 7:323–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin Z, Ogata S, Tamura G, Katayama Y,
Fukase M, Yajima M and Motoyama T: Carcinosarcomas (malignant
mullerian mixed tumors) of the uterus and ovary: A genetic study
with special reference to histogenesis. Int J Gynecol Pathol.
22:368–373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Icard P, Goudet P, Charpenay C,
Andreassian B, Carnaille B, Chapuis Y, Cougard P, Henry JF and
Proye C: Adrenocortical carcinomas: Surgical trends and results of
a 253-patient series from the French Association of Endocrine
Surgeons study group. World J Surg. 25:891–897. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hutter AM Jr and Kayhoe DE: Adrenal
cortical carcinoma. Clinical features of 138 patients. Am J Med.
41:572–580. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bourdeau I, MacKenzie-Feder J and Lacroix
A: Recent advances in adrenocortical carcinoma in adults. Curr Opin
Endocrinol Diabetes Obes. 20:192–197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fleming S: Carcinosarcoma (mixed
mesodermal tumor) of the ureter. J Urol. 138:1234–1235.
1987.PubMed/NCBI
|
|
25
|
Beasley MB, Brambilla E and Travis WD: The
2004 World Health Organization classification of lung tumors. Semin
Roentgenol. 40:90–97. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Craig JR, Peters RL and Edmondson HA:
Tumors of the Liver and Intrahepatic Bile Ducts. Armed Forces
Institute of Pathology (Washington, DC). 185–186. 1989.
|
|
27
|
Koss MN, Hochholzer L and Frommelt RA:
Carcinosarcomas of the lung: A clinicopathologic study of 66
patients. Am J Surg Pathol. 23:1514–1526. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dackiw AP, Lee JE, Gagel RF and Evans DB:
Adrenal cortical carcinoma. World J Surg. 25:914–926. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Feng YC, Yang ZG, Chen TW, Su XY, Deng W
and Wang QL: Adrenal sarcomatoid carcinoma: A rare case depicted on
multi-detector row computed tomography. Indian J Med Sci. 64:37–40.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van't Sant HP, Bouvy ND, Kazemier G,
Bonjer HJ, Hop WCJ, Feelders RA, de Herder WW and de Krijger RR:
The prognostic value of two different histopathological scoring
systems for adrenocortical carcinomas. Histopathology. 51:239–245.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kendrick ML, Lloyd R, Erickson L, Farley
DR, Grant CS, Thompson GB, Rowland C, Young WF Jr and van Heerden
JA: Adrenocortical carcinoma: Surgical progress or status quo? Arch
Surg. 136:543–549. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schteingart DE, Doherty GM, Gauger PG,
Giordano TJ, Hammer GD, Korobkin M and Worden FP: Management of
patients with adrenal cancer: Recommendations of an international
consensus conference. Endocr Relat Cancer. 12:667–680. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Terzolo M, Daffara F, Ardito A, Zaggia B,
Basile V, Ferrari L and Berruti A: Management of adrenal cancer: A
2013 update. J Endocrinol Invest. 37:207–217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kamio T, Shigematsu K, Sou H, Kawai K and
Tsuchiyama H: Immunohistochemical expression of epidermal growth
factor receptors in human adrenocortical carcinoma. Hum Pathol.
21:277–282. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang X, MacLennan GT, Zhang S, Montironi
R, Lopez-Beltran A, Tan PH, Foster S, Baldridge LA and Cheng L:
Sarcomatoid carcinoma of the upper urinary tract: Clinical outcome
and molecular characterization. Hum Pathol. 40:211–217. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Italiano A, Cortot AB, Ilie M,
Martel-Planche G, Fabas T, Pop D, Mouroux J, Hofman V, Hofman P and
Pedeutour F: EGFR and KRAS status of primary sarcomatoid carcinomas
of the lung: Implications for anti-EGFR treatment of a rare lung
malignancy. Int J Cancer. 125:2479–2482. 2009. View Article : Google Scholar : PubMed/NCBI
|