Introduction
There are numerous microdomain-mediated
intracellular signaling pathways that regulate cellular functions
and act as selective transduction mediators that control
interactions between internal and external environments of cells
(1). The insulin-like growth factor 1
(IGF-1)/IGF-1 receptor (IGF-1R) system plays important roles in the
carcinogenesis and progression of gastrointestinal (GI) cancers
(2). After the ligands bind to the
external subunit of the IGF-1R, a conformational change is induced
in the trans-membrane β subunits, resulting in autophosphorylation
of cytoplasmic tyrosine kinase. IGF-1R subsequently phosphorylates
intracellular substrates, including insulin receptor substrates 1
to 4 and Shc (2). These early events
activate multiple signaling pathways, including the
mitogen-activated protein kinase (MAPK), extracellular
signal-regulated kinase (ERK) and phosphoinositide 3-kinase
(PI3-K)/Akt-1 pathways (3,4). These pathways then switch on several
cellular functions, including anti-apoptosis, transcription,
metabolism, proliferation and growth.
In normal cells, the IGF-1/IGF-1R system is
regulated by multiple steps (5). The
expression of growth hormone (GH) is activated by GH-releasing
hormone (GHRH). GH then increases the secretion of IGFs and
IGF-binding proteins (IGFBPs) from hepatocytes. Activation of
IGF-1R is tightly regulated by the amount of the free form of its
ligand. IGFBPs 1 to 6 circulate and modulate IGF activity by
reducing the bioavailability of IGF to bind to the IGF-1R (2) Dysregulation of the IGF/IGF-1R system has
been implicated in the proliferation of numerous tumors, such as
gastric, pancreas, esophageal and colorectal cancer (6,7). Exogenous
IGF stimulates the proliferation of various cancer cells. In
addition, overexpressed IGF-1R signals are also important in tumor
dissemination through the control of adhesion, migration and
metastasis (8,9). There is known to be a positive feedback
loop between the IGF/IGF-1R axis and matrilysin for the progression
and invasion of GI cancers (2).
Therefore, blocking IGF-1R inhibits tumor progression through
several interruptions of IGF-1R-mediated functions, including the
IGF-1R and matrilysin positive feedback system (2).
By contrast, statins are extremely popular drugs for
lowering serum cholesterol levels by inhibiting
3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase.
Previously, statins have been studied for their pleiotropic
effects, including anti-inflammatory, anti-oxidant and anti-cancer
effects. Statins reduce serum cholesterol levels by competitively
inhibiting HMG-CoA reductase, the rate-limiting enzyme in the
mevalonate pathway. Mevalonate, in addition to being involved in
cholesterol synthesis, is also involved in the synthesis of
isoprenyl proteins, dolichols and ubiquinone, which perform several
key cellular functions, such as cell signaling, proliferation,
growth and respiration. Certain previous studies have shown statins
to be beneficial as anti-cancer drugs (10,11). The
antitumor effects of statins may be due to the inhibition of cell
proliferation, promotion of apoptosis, inhibition of angiogenesis
and prevention of metastasis.
Therefore, the present study investigated whether
simvastatin induces apoptosis of human colon cancer cells and how
statin affects IGF-1R and its associated signaling pathways in
colon cancer cells.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), trypsin, EDTA, penicillin and streptomycin were
from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Pravastatin and simvastatin were from Calbiochem (Merck Millipore,
Darmstadt, Germany). Human IGF-1 was from R&D Systems, Inc.
(Minneapolis, MN, USA). Rabbit polyclonal antibodies against p44/42
MAPK (Erk1/2; #9102), phospho-p44/42 MAPK (Erk1/2; Thr202/Tyr204;
#9101), Akt (#9272) and phospho-Akt (Ser473; #9271) were from Cell
Signaling Technology, Inc. (Danvers, MA, USA). β-actin
(#sc-130656), rabbit polyclonal anti-IGF-1R (#sc-713) and goat
anti-rabbit immunoglobulin G (IgG)-horseradish peroxidase (HRP;
#sc-2768) antibodies were from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Amersham ECL Advance Western Blotting Detection
kit was from GE Healthcare Life Sciences (Chalfont, UK).
Cell culture
Human HT-29 cells were provided by Dr C. S. Eun
(Hanyang University College of Medicine, Seoul, Korea) and were
cultured in DMEM with 4.5 g/l glucose and 2 mM glutamine
supplemented with 10% FBS, 1.5 g/l sodium bicarbonate, 100 IU/ml
penicillin and 100 µg/ml streptomycin. The medium was changed twice
a week and the cells were maintained in an incubator at 37°C with a
5% CO2 atmosphere. The cells were subcultured when
confluent (every 5–7 days) using trypsin (2.5 g/l) and EDTA (1
g/l). Experiments were performed in serum-free medium containing
0.1% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO,
USA).
Methyl thiazolyl tetrazolium (MTT)
assay
Cell proliferation of HT-29 cells was measured using
an MTT assay. The HT-29 cells were seeded with DMEM culture medium
at a density of 5×104 cells/ml in a 96-well plate.
Following incubation at 37°C for 24 h, the cells were treated with
various concentrations of pravastatin or simvastatin (2.5–20 µM) in
serum-free medium for 24 or 48 h. MTT (0.5 mg/ml; Sigma-Aldrich)
was then added to each well and incubated for an additional 4 h at
37°C. Subsequent to removal of the medium, 100 µl dimethyl
sulfoxide was added to each well, and the plates were weakly
agitated for 10 min. The optical density (OD) was evaluated by DTX
880 Multimode Detector (Beckman Coulter, Brea, CA, USA) at 570 nm.
Each assay was performed in triplicate.
Cell cycle analysis
The HT-29 cells were seeded with the DMEM culture
medium at a concentration of 1×104 cells/well into a
6-well plate and incubated for 24 or 48 h. Cells were treated with
simvastatin (50 µM) for 48 or 72 h. The cells were harvested with
0.25% trypsin-EDTA and washed with PBS. The cells were centrifuged
twice at 1,400 × g for 5 min and then fixed with in 1 ml of 70%
ethanol at −20°C for 1 h. Subsequent to centrifugation at 1,400 × g
for 5 min, the supernatant was removed and cells were incubated
with 1 ml PBS containing 10 µl RNase (10 µg/ml; Sigma-Aldrich) and
20 µl propidium iodide (1 mg/ml; Sigma-Aldrich) at 37°C for 1 h.
The cell cycle was analyzed using the FACSCalibur flow cytometer
and CellQuest software (BD Biosciences, San Jose, CA, USA). Flow
cytometry was used to determine the percentage of cells in each
phase of the cell cycle.
Cell death assay
Cell apoptosis was assessed by the detection of
mono-oligonucleosomes, which are histone-associated DNA fragments,
using a Cell Death Detection ELISAPLUS kit
(#11774425001; Roche Applied Science, Mannheim, Germany), according
to the manufacturer's protocol. The HT-29 cells were seeded with
the culture medium in a 96-well plate at a concentration of
1×104 cells/well and incubated at 37°C for 24 h. The
cells were treated with various concentrations (2.5–20 µM) of
pravastatin or simvastatin for 24 or 48 h. Subsequent to removal of
the medium, the cells were treated with 100 µl lysis buffer for 30
min and centrifuged at 200 × g at 4°C for 10 min. The supernatant,
which consisted of the cell lysate solution, was placed in the
streptavidin-coated microplate supplied with the kit. A mixture of
the provided anti-histone-biotin and anti-DNA-POD antibodies were
added to the cell lysate and incubated for 2 h. Subsequent to
washing the plate, 100 µl of
2,2′-azinobis-3-ethyl-benzothiazoline-6-sulfonic acid substrate was
added to each well of the plate for 20 min. The absorbance at 405
nm was measured using a DTX 880 Multimode Detector (Beckman
Coulter).
Caspase-3 activity assay
Caspase-3 activity assay (BioVision, Mountain View,
CA, USA) was used to measure caspase-3 activity, according to the
manufacturer's protocol. The cells were plated on 60 mm dishes in
culture medium at a concentration of 2×106 cells/ml and
treated with various concentrations of pravastatin or simvastatin
(5–50 µM) for 48 h. Cells were washed with PBS and harvested with
lysis buffer. Cells were kept on ice for 10 min. The cell lysate
was centrifuged at 12,000 × g at 4°C and supernatant was
transferred to a new tube and stored on ice. Protein contents were
analyzed using the Bradford assay (Sigma-Aldrich). Assays were
performed in 96-well plates containing 90 µg of protein in 50 µl
lysis buffer, and 5 µl of 4 mM DEVD-pNA was added to the protein
samples. The samples were incubated at 37°C for 2 h. Absorbance was
measured at 405 nm using the DTX 880 Multimode Detector.
Western blotting
The cells were washed with PBS and harvested with
radioimmunoprecipitation assay lysis buffer containing 50 mM Tris
(pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium
deoxycholate, 0.1% SDS, 1 µM phenylmethylsulfonyl fluoride, 5 µg/ml
aprotinin and 5 µg/ml leupeptin. Protein contents were analyzed
using the Bradford assay (Sigma-Aldrich). Sodium dodecyl
sulfate-polyacrylamide gel electrophoresis was performed with a 4%
stacking gel and a 10% resolving gel, followed by transfer to
nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The membranes were blocked for 1 h at room temperature in
blocking solution containing 5% skim milk in Tris-buffered saline
with Tween-20 (TBS-T), which consisted of 200 mM Tris, 500 mM NaCl
(pH 7.5) and 0.05% v/v Tween-20 (Sigma-Aldrich). The membranes were
then incubated overnight at 4°C in 5% BSA solution, consisting of
5% BSA in TBS-T, with the phosphorylated-p44/42 MAPK,
non-phosphorylated-p44/42 MAPK, phosphorylated-Akt,
non-phosphorylated-Akt or IGF-1R antibodies (dilution, 1:1,000).
The membranes were washed with TBS-T and incubated with a goat
anti-rabbit IgG-HRP antibody for 1 h at room temperature. The
membranes were washed and developed using an Amersham ECL Advance
Western Blotting Detection kit for 2 min, and autoradiography was
performed. The signal intensities for specific bands on the western
blots were semi-quantified using ImageJ density analysis software
(Version 1.43; National Institutes of Health, Bethesda, MA,
USA).
Statistical analysis
Results from each experiment were expressed as the
mean ±standard deviation of three separate experiments. Data were
analyzed by one-way analysis of variance and by Tukey's multiple
comparisons test using GraphPad Prism software version 4.0
(GraphPad Software, Inc., La Jolla, CA, USA).
Results
Simvastatin suppresses cell
proliferation in HT-29 cells
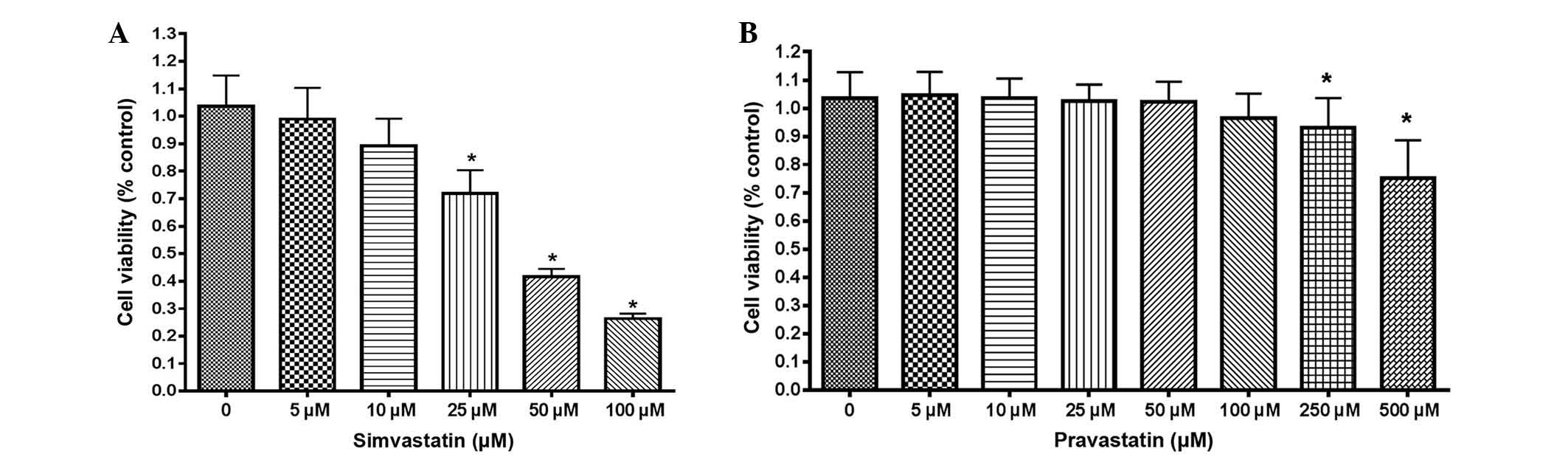
The proliferation of HT-29 cells was first examined
by statins using an MTT assay. Simvastatin suppressed the
proliferation of HT-29 cells in a dose-dependent manner (Fig. 1A). Cell proliferative activity was
significantly suppressed at doses >25 µM simvastatin, compared
with untreated control cells (P<0.001). However, HT-29 cells
were more resistant to treatment with pravastatin than simvastatin,
since the viability of HT-29 cells treated with pravastatin was
significantly reduced at a dose of 250 µM, while the viability of
cells treated with simvastatin was reduced at 25 µM (Fig. 1B).
Simvastatin induces apoptosis via
caspase-3 activation, downregulates the expression of B cell
lymphoma-2 (Bcl-2) and upregulates the expression of
Bcl-2-associated X protein (Bax)
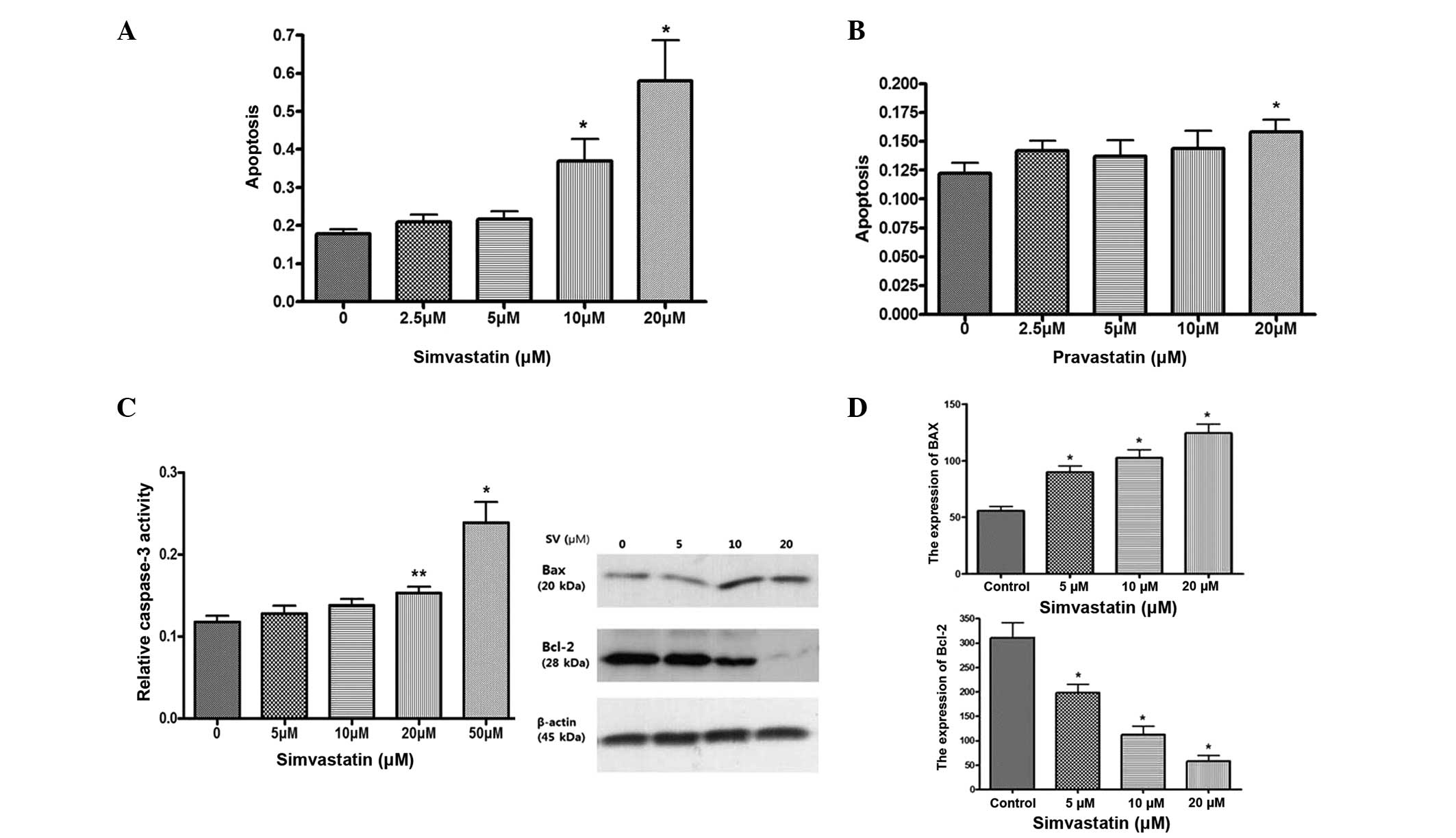
The effect of statins on cell death was investigated
using a cell death detection ELISA assay. Simvastatin significantly
induced apoptosis in HT-29 cells at a dose of >10 µM
simvastatin, compared with control cells, while pravastatin was not
as effective as simvastatin for inducing cell death of HT-29 cells
(Fig. 2A and B). Simvastatin also
significantly increases caspase-3 activity in a dose dependent
manner (Fig. 2C). Western blotting of
the proapoptotic protein Bax and anti-apoptotic protein Bcl-2 was
also examined. Simvastatin significantly upregulated the expression
of Bax and downregulated the expression of Bcl-2 (Fig. 2D). These results suggest that
simvastatin induces apoptosis of HT-29 cells.
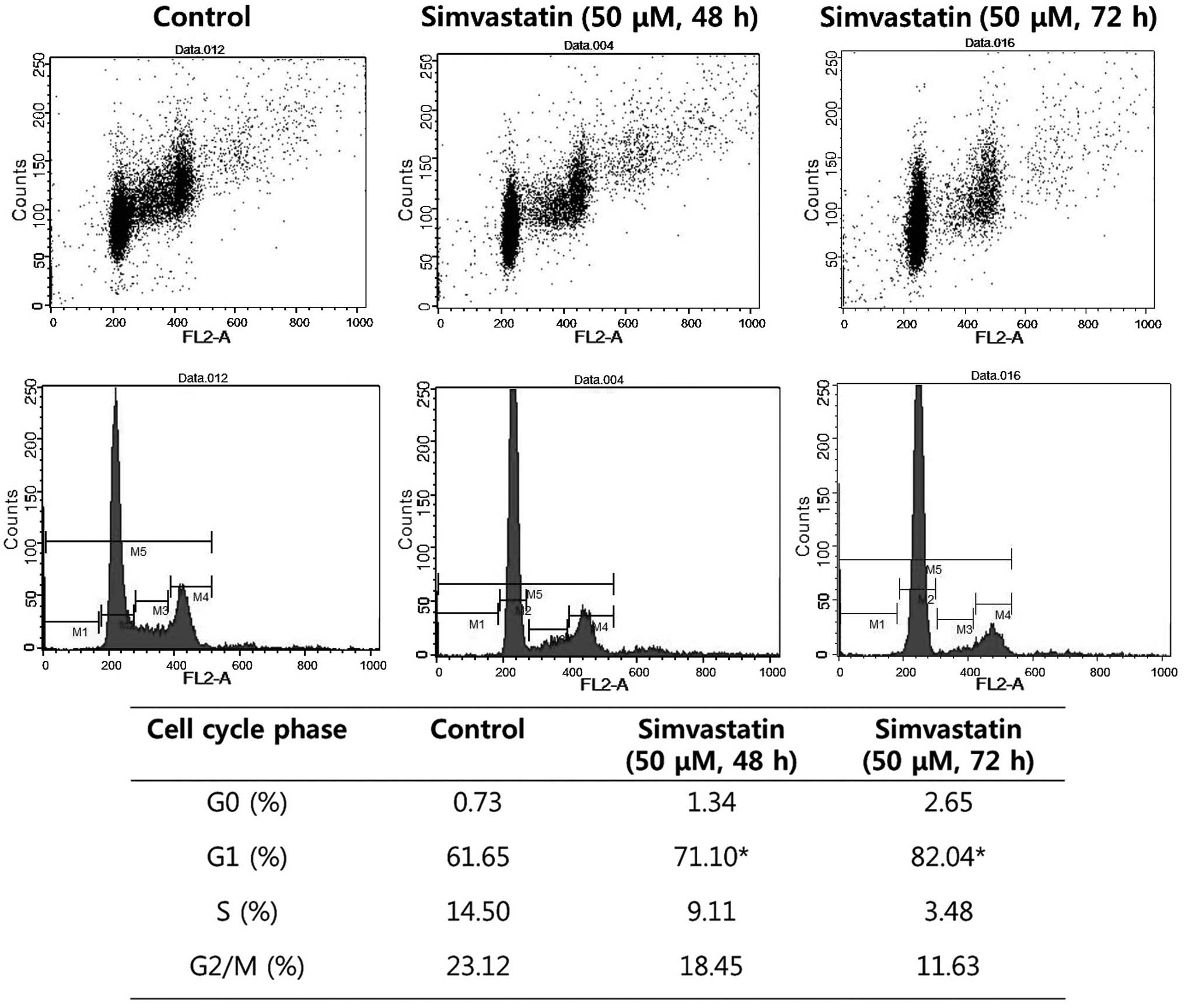
Simvastatin induces G1-phase cell
cycle arrest
Cell cycle analysis by flow cytometry was performed
on HT-29 cells subsequent to 48 and 72 h treatment with
simvastatin. The cell population in the G1 phase was increased by
9.45 and 20.39%, respectively, compared with the control
(P<0.05). These results demonstrated that simvastatin induced
the arrest of colon cancer cells in the G1 phase (Fig. 3).
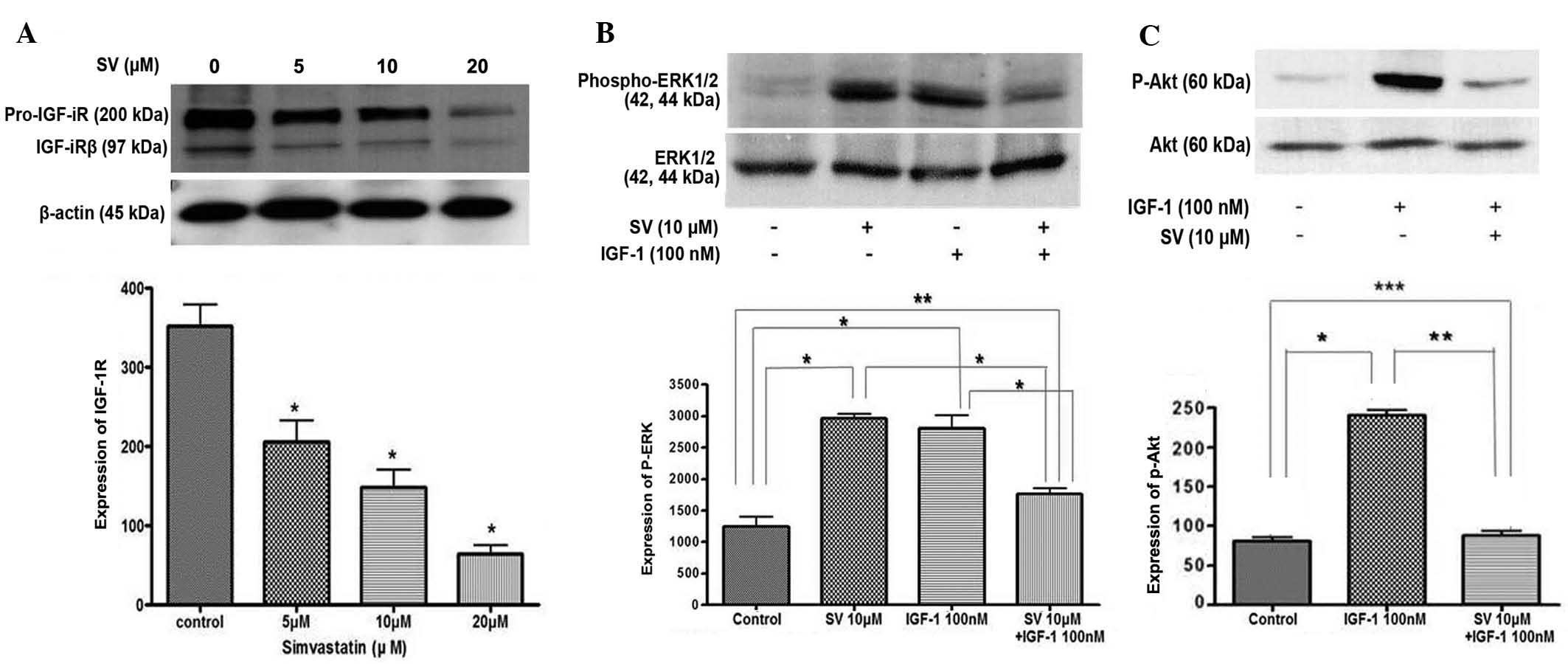
Simvastatin suppresses the expression
of IGF-1R and IGF-1-induced ERK/Akt activation
The present study examined how simvastatin affects
IGF-1R and its signaling pathways in HT-29 cells. Treatment with
simvastatin significantly downregulated the expression of IGF-1Rβ
in a dose-dependent manner, compared with the untreated control
cells (Fig. 4A). In addition, the
present study examined the effect of simvastatin on the IGF-1R
signaling pathway. HT-29 cells were pretreated with 10 µM
simvastatin for 24 h and then stimulated with 100 nM IGF-1 for 15
min. IGF-1 treatment alone stimulated ERK phosphorylation, which
was significantly suppressed by simvastatin treatment. However, 10
µM simvastatin alone also induced phosphorylated ERK activation in
a similar manner to IGF-1 treatment (Fig.
4B).
For the next step, the same experiment was performed
to examine Akt activation. First, IGF-1 induced Akt
phosphorylation. IGF-1-induced phosphorylated Akt was also
significantly suppressed subsequent to simvastatin treatment
(Fig. 4C). These results indicate
that simvastatin suppresses IGF-1-induced ERK and Akt activation in
HT-29 cells.
Simvastatin induced proapoptotic ERK
activation and antagonized IGF-1-induced anti-apoptotic ERK
To understand why ERK was activated by IGF-1 and
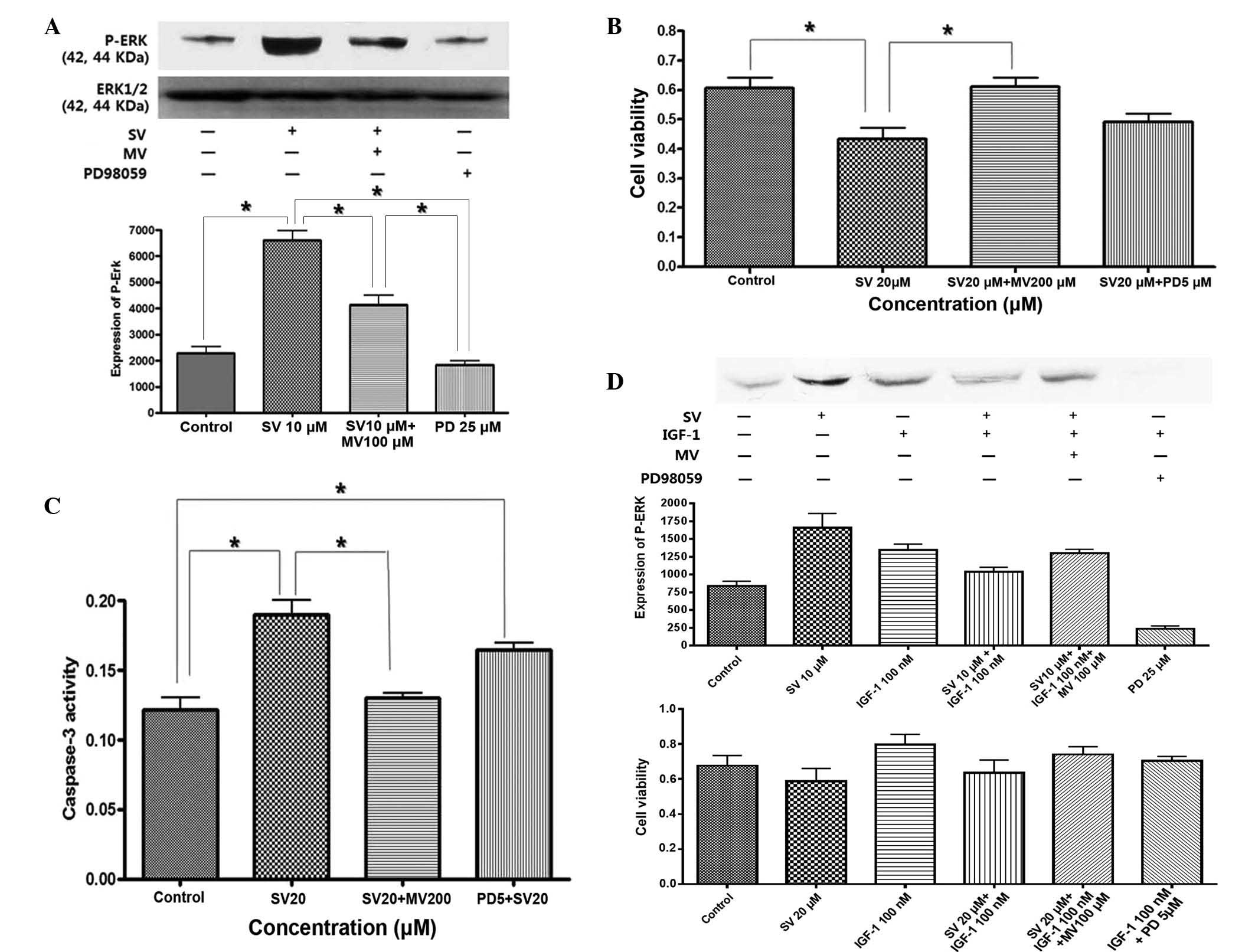
simvastatin, the effect of mevalonic acid on the MAPK pathway was
investigated. ERK activity was blocked with the MEK inhibitor
PD98059. ERK activity and the degree of cell proliferation was then
evaluated. As shown in Fig. 5A,
treatment with simvastatin for 24 h stimulated sustained activation
of phosphorylated ERK, which was reversed by treatment with
mevalonic acid and PD98059. Apoptosis was induced by treatment with
simvastatin, but this was fully reversed by pretreatment with
mevalonic acid and partially reversed by pretreatment with PD98059
(Fig. 5B and C). This result
indicates that simvastatin induced apoptosis through selective and
sustained activation of proapoptotic ERK.
By contrast, IGF-1 induced phosphorylation of ERK,
which peaked at 15 min subsequent to treatment with IGF-1, and led
to cell proliferation, while simvastatin suppressed IGF-1-induced
ERK activation and cell proliferation, which was also reversed by
pretreatment with mevalonic acid (Fig.
5D). This result indicated that IGF-1 stimulated anti-apoptotic
ERK phosphorylation, while simvastatin induced proapoptotic ERK
activation and also antagonized IGF-1-induced anti-apoptotic ERK
activation through sustained ERK phosphorylation.
Discussion
In the present study, it was demonstrated that
simvastatin inhibited the proliferation of colon cancer cells and
suppressed the expression of IGF-1R and IGF-1-induced ERK/Akt
activation in HT-29 cells. To the best of our knowledge, the
present study showed for the first time that simvastatin induced
proapoptotic ERK and antagonized IGF-1-induced anti-apoptotic ERK
in colon cancer cells.
IGF-1R is synthesized as a single precursor peptide
that is cleaved into the α and β chains, and is transported to the
membrane fully assembled in the dimeric form, with two α chains and
two β subunits. Subsequent to transportation, the ligands bind to
the subunits of IGF-1R, which is autophosphorylated to stimulate
tyrosine kinase in the intracellular domain of the β subunits
(2–4).
These early events activate multiple signaling pathways, including
the MAPK, ERK and PI3-K/Akt-1 pathways. Overexpression of IGF-IR
has been shown in numerous solid cancers and is associated with
cancer progression and metastasis (12,13). It
has been reported that IGF-IR is overexpressed or over-activated in
almost 80% of colon cancers (14,15). In
the present study, statins induced downregulation of the β subunits
of IGF-1R and suppressed the subsequent ERK and PI3-K/Akt
activation induced by IGF-1 in HT-29 cells. These results indicate
that statins could control the IGF-1R signaling pathway, which is
important in the growth and metastasis of colon cancer.
Statins cause substantial reduction in the serum
cholesterol level, other downstream mevalonate products (such as
isoprenoid molecules, farnesyl pyrophosphate and
geranylgeranylpyrophosphate, which are necessary for
post-translational modification) and isoprenylation of a variety of
proteins that are involved in cell motility, such as Ras and Rho
(11,16). Therefore, statins are considered to be
plausible treatment options as lowering the cholesterol content,
including cell structural and functional elements, may inhibit
cancer proliferation (11,16,17).
Several studies have investigated statin-attenuated IGF-1-induced
ERK activation and cell proliferation in mesangial cells and
prostate cancer cells (16,17). One study demonstrated that fluvastatin
inhibited IGF-1-induced MEK1/2 and ERK1/2 phosphorylation,
mesangial cell proliferation and cyclin D1 expression (16). In another study, simvastatin was found
to suppress proliferation and induce apoptosis of prostatic cancer
cells, and to suppress the expression of IGF-1R. Knockdown of
IGF-1R by siRNA resulted in the inhibition of proliferation of
prostatic cancer cells. Simvastatin also inhibited IGF-1-induced
activation of both ERK and Akt signaling and IGF-1-induced
prostatic cancer cell proliferation (17). In another previous study, knockdown of
IGF-1R by RNA interference in the colon cancer SW480 cell line
suggested that decreasing the IGF-1R protein level could
significantly inhibit tumor growth (18). These studies supported the present
results that statin is a potent inhibitor of IGF-1R signaling
system in colon cancer.
Notably, simvastatin induced ERK activation in
addition to inhibiting IGF-1-induced ERK activation in HT-29 cells.
The ERK1/2 cascade transmits mostly mitogenic signals and regulates
growth factor-induced cell proliferation (19). The present results suggested that ERK
may act differently, depending on the types of stimulating agents
and the duration or strength of the signal. As a similar
experimental result for time-dependent response, epidermal growth
factor (EGF) and nerve growth factor (NGF) stimulate strong
activation of ERK1/2 with distinct outcomes (20). EGF stimulation caused a transient
activation of ERK1/2, peaking at 15 min and reducing back to basal
levels after 40 min, and induced cell proliferation in PC12 cells,
while NGF stimulated sustained activation of ERK1/2 after 15–180
min, which resulted in the differentiation of cells (20). Previously, β,β-dimethylacrylshikonin
induced mitochondria-dependent apoptosis through the ERK pathway in
human gastric cancer SGC-7901 cells (21). Shen et al (21) suggested that sustained ERK activation
induces apoptosis. In this study, a specific inhibitor of MEK
blocked β,β-dimethylacrylshikonin-induced ERK activation and
apoptosis in SGC-7901 cells (21).
Kim et al showed that glucose deprivation induced adenosine
monophosphate-activated kinase (AMPK) and proapoptotic ERK
activation in HCT116 cells (22).
AMPK protected HCT116 cells from glucose deprivation by suppressing
proapoptotic ERK. From these results, it was found that ERK
activation leads to cell proliferation, differentiation and
apoptosis under various stimuli in various tissues.
Overall, the present study found that simvastatin
induces apoptosis and suppresses the β subunits of IGF-1R and
IGF-1-induced ERK/Akt activation. The present results also
demonstrated that simvastatin induces proapoptotic ERK activation,
which antagonizes IGF-1-induced anti-apoptotic ERK activation in
HT-29 cells. These results suggested that statins may be potential
anti-cancer drugs against colon cancer due to proapoptotic ERK
control.
Acknowledgements
The present study was supported by the Hallym
University Medical Center (grant no. 01-2011-20).
References
|
1
|
Jahn KA, Su Y and Braet F: Multifaceted
nature of membrane microdomains in colorectal cancer. World J
Gastroenterol. 17:681–990. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Adachi Y, Yamamoto H, Ohashi H, Endo T,
Carbone DP, Imai K and Shinomura Y: A candidate targeting molecule
of insulin-like growth factor-I receptor for gastrointestinal
cancers. World J Gastroenterol. 16:5779–5789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baserga R: The insulin-like growth factor
I receptor: A key totumor growth? Cancer Res. 55:249–252.
1995.PubMed/NCBI
|
|
4
|
Yu H and Rohan T: Role of the insulin-like
growth factor family in cancer development and progression. J Natl
Cancer Inst. 92:1472–1489. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodon J, De Santos V, Ferry RJ Jr and
Kurzrock R: Early drug development of inhibitors of the
insulin-like growth factor-Ireceptor pathway: Lessons from the
first clinical trials. Mol Cancer Ther. 7:2575–2588. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen W, Wang S, Tian T, Bai J, Hu Z, Xu Y,
Dong J, Chen F, Wang X and Shen H: Phenotypes and genotypes of
insulin-like growth factor 1, IGF-binding protein-3 and cancer
risk: Evidence from 96 studies. Eur J Hum Genet. 17:1668–1675.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rinaldi S, Cleveland R, Norat T, Biessy C,
Rohrmann S, Linseisen J, Boeing H, Pischon T, Panico S, Agnoli C,
et al: Serum levels of IGF-I, IGFBP-3 and colorectal cancer risk:
Results from the EPIC cohort, plus a meta-analysis of prospective
studies. Int J Cancer. 126:1702–1715. 2010.PubMed/NCBI
|
|
8
|
Dunn SE, Kari FW, French J, Leininger JR,
Travlos G, Wilson R and Barrett JC: Dietary restriction reduces
insulin-like growth factor I levels, which modulates apoptosis,
cell proliferation, and tumor pogression in p53-deficient mice.
Cancer Res. 57:4667–4672. 1997.PubMed/NCBI
|
|
9
|
Wu Y, Yakar S, Zhao L, Hennighausen L and
LeRoith D: Circulating insulin-like growth factor-I levels regulate
colon cancer growth and metastasis. Cancer Res. 62:1030–1035.
2002.PubMed/NCBI
|
|
10
|
Blais L, Desgagne´ A and LeLorier J:
3-Hydroxy-3-methylglutaryl coenzymeA reductase inhibitors and the
risk of cancer: A nested case-control study. Arch Intern Med.
160:2363–2368. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Graaf MR, Beiderbeck AB, Egberts AC,
Richel DJ and Guchelaar HJ: The risk of cancer in users of statins.
J Clin Oncol. 22:2388–2394. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pollak M: Insulin and insulin-like growth
factor signalling in neoplasia. Nat Rev Cancer. 8:915–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gallagher EJ and LeRoith D: The
proliferating role of insulin and insulin-like growth factors in
cancer. Trends Endocrinol Metab. 21:610–618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reinmuth N, Fan F, Liu W, Parikh AA,
Stoeltzing O, Jung YD, Bucana CD, Radinsky R, Gallick GE and Ellis
LM: Impact of insulin-like growth factor receptor-I function on
angiogenesis, growth, and metastasis of colon cancer. Lab Invest.
82:1377–1389. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
LeRoith D and Roberts CT: The insulin-like
growth factor system and cancer. Cancer Lett. 195:127–137. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shibata T, Tamura M, Kabashima N, Serino
R, Tokunaga M, Matsumoto M, Miyamoto T, Miyazaki M, Furuno Y,
Takeuchi M, et al: Fluvastatin attenuates IGF-1-induced ERK1/2
activation and cell proliferation by mevalonic acid depletion in
human mesangial cells. Life Sci. 84:725–731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sekine Y, Furuya Y, Nishii M, Koike H,
Matsui H and Suzuki K: Simvastatin inhibits the proliferation of
human prostate cancer PC-3 cells via down-regulation of the
insulin-like growth factor 1 receptor. Biochem Biophys Res Commun.
372:356–361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yavari K, Taghikhani M, Maragheh MG,
Mesbah-Namin SA and Babaei MH: Knockdown of IGF-IR by RNAi inhibits
SW480 colon cancer cells growth in vitro. Arch Med Res. 40:235–240.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wortzel I and Seger R: The ERK cascade:
Distinct functions within various subcellular organelles. Genes
Cancer. 2:195–209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chung J, Kubota H, Ozaki Y, Uda S and
Kuroda S: Timing-dependent actions of NGF required for cell
differentiation. PLoS One. 5:e90112010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen XJ, Wang HB, Ma XQ and Chen JH:
β,β-Dimethylacrylshikonin induces mitochondria dependent apoptosis
through ERK pathway in human gastric cancer SGC-7901 cells. PLoS
One. 7:e417732012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim MJ, Park IJ, Yun H, Kang I, Choe W,
Kim SS and Ha J: AMP-activated protein kinase antagonizes
pro-apoptotic extracellular signal-regulated kinase activation by
inducing dual-specificity protein phosphatases in response to
glucose deprivation in HCT116 carcinoma. J Biol Chem.
285:14617–14627. 2010. View Article : Google Scholar : PubMed/NCBI
|