Introduction
Glucose-regulated protein 78 kDa/binding
immunoglobulin protein (GRP78/BIP) is an endoplasmic reticulum (ER)
chaperone protein and a member of the heat shock protein (Hsp) 70
family. GRP78 has critical roles in protein homeostasis and the
unfolded protein response. It has been identified in numerous
organisms from yeast to humans (1–4). The
expression of GRP78 clearly increases during ER stress, and
therefore is used as an ER stress marker (4). In addition, GRP78 is involved in the
integrity and the regulation of the ER, and it prevents the
aggregation of misfolded proteins, including apoB100, by targeting
them to the degradation by the 26S proteasome (5). When ER stress occurs, GRP78 is separated
from inositol-requiring enzyme 1, protein kinase RNA-like
endoplasmic reticulum kinase and/or activating transcription factor
6, thus enabling them to be activated, which leads to the unfolded
protein response, cell survival or apoptosis (4).
Studies have revealed that GRP78 is overexpressed in
a wide range of tumors and therefore may be responsible for the
resistance to hormonal therapies as well as chemotherapeutics.
Zhang et al (6) previously
demonstrated that breast and prostate cancer cells resistant to
hormonal therapy actively promote GRP78 to the cell surface. In
addition, that study revealed that soluble GRP78 forms a complex
with phosphoinositide 3-kinase (PI3K), leading to PI3K activation,
which is known be activated in various cancer cells resulting in
proliferation and therapeutic resistance (6). In addition, GRP78 forms were identified
as mediating resistance to ionizing radiation in a stem cell-like
subpopulation within the human breast cancer MCF-7 cell line
(7). Furthermore, when GRP78 is
suppressed by lentiviral vectors expressing small interfering (si)
RNA, human breast cancer cells become sensitized to
etoposide-mediated cell death (8).
Dong et al (8) demonstrated
that treatment with combretastatin A4P, a vascular targeting agent,
or contortrostatin, an antiangiogenic agent, promoted
transcriptional activation of the GRP78 promoter and increased
GRP78 protein in MDA-MB-435 xenografts tumor cells. Additionally,
the level of GRP78 expression in primary tumors from resected
gastric cancer and metastatic lymph nodes was inversely associated
with patient survival (9). Zhang
et al (9) demonstrated that
knocking down GRP78 expression inhibits tumor cell invasion, growth
and metastasis in a xenograft nude mouse model; therefore, leading
to the conclusion that a dysregulated expression of GRP78 may
contribute to the development and progression of gastric
cancer.
Wang et al (10) reported that GRP78 expression appears
to be critical to the responsiveness of proteasomal inhibition in
various thyroid cancer cell lines; it was demonstrated that
insensitive thyroid cancer cell lines are sensitized to the
proteasome inhibition by suppression of GRP78. The 26S proteasome
inhibitor bortezomib, also known as Velcade or PS-341, interferes
with ER responses and improves survival of patients with aggressive
hematologic malignant tumors (11).
siRNA silencing of GRP78 renders diffuse large B-cell lymphoma
(DLBCL) cell lines sensitive to bortezomib (11). Chen et al (12) demonstrated that exposure of 9l rat
brain cells to low concentrations of thapsigargin (TG), a
sarcoendoplasmic Ca2+-ATPase inhibitor, leads to
immediate suppression of general protein synthesis and enhanced
induction of GRP78. Those authors also revealed that TG-induced
GRP78 expression may be suppressed by cytosolic free calcium
(Ca2+c) chelator dibromo-1,2-bis
(aminophenoxy) ethane N, N, N', N'-tetraacetic acid (BAPTA), which
enters cells as an ester derivative BAPTA-acetoxymethyl ester
(BAPTA-AM), and the induction of GRP78 expression was completely
inhibited in the presence of 20 µM BAPTA-AM (12). Mozos et al (11) demonstrated that reducing GRP78
expression by treating bortezomib-resistant DLBCL cell lines with
prednisone overcomes bortezomib resistance.
The current study examined the expression of GRP78
in response to bortezomib-treatment in the highly metastatic mouse
breast cancer 4T1 cell line. The present results revealed that
GRP78 is significantly induced in a dose- and time-dependent manner
following low doses of bortezomib-treatment. In addition, the
results demonstrated that combination treatment with bortezomib and
the intracellular calcium chelator BAPTA-AM may be a novel
treatment strategy for breast cancer.
Materials and methods
Materials
RPMI-1640 media, fetal bovine serum (FBS),
penicillin/streptomycin,
3-(4,5-dimethylthylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
(MTT), leupeptin and BAPTA-AM were obtained from Sigma-Aldrich (St.
Louis, MO, USA). Rabbit polyclonal anti-GRP78 and anti-ubiquitin
antibodies were obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX. USA), and rabbit polyclonal anti-β-actin antibody was
obtained from Abcam (Cambridge, MA, USA). Cell Proliferation
Reagent WST-1 was obtained from Roche Diagnostics GmbH (Mannheim,
Germany). Bortezomib was generously provided by Dr Engin Ulukaya
(Uludağ University, Bursa, Turkey). All other reagents were
purchased from Sigma-Aldrich unless otherwise specified.
Cell culture
Mouse breast cancer 4T1 cell line (obtained from Dr
Nuray Erin, Akdeniz University, Antalya, Turkey) was cultured in
RPMI-1640 (plus 4.5 g/l glucose, 10 mM HEPES, 1 mM sodium pyruvate,
0.15% sodium bicarbonate, 100 µg/ml streptomycin and 100 U/ml
penicillin) containing 10% FBS. Cell cultures were maintained at
37°C in a 5% CO2 humidified incubator in 25
cm2 Corning flasks (Corning Incorporated, Corning, NY,
USA). The cells were subcultured at ~70% confluency.
Western blot analysis
4T1 cells (100,000 per 35×10 mm petri dishes) were
treated with dimethyl sulfoxide (DMSO; control), 10 µM leupeptin or
various concentrations of bortezomib (10–200 nM) for 24 h. For
time-dependent experiments, the cells were treated with 10 nM
bortezomib for 12, 24 and 48 h. After treatment, the cells were
lyzed with radioimmunoprecipitation assay lysis buffer containing
protease inhibitor cocktail. A total of 25 or 40 µg protein from
each sample was separated by 12% sodium dodecyl sulfate (SDS)
polyacrylamide gel electrophoresis. Subsequently, the proteins were
transferred to polyvinylidene difluoride membranes at 70 V for 2 h.
Subsequently, the membranes were blocked with 5% non-fat dried milk
in Tris-buffered saline with Tween 20 (TBS-T). The membranes were
then incubated with GRP78 antibody (1:500; catalog no., sc-13968),
ubiquitin antibody (1:200; catalog no., sc-9133) or β-actin
antibody (1:500; catalog no., ab8227) in TBS-T for 1 h. The
membranes were incubated with donkey anti-rabbit horseradish
peroxidase (HRP)-conjugated secondary antibody (1:5,000; catalog
no., NIF824; GE Healthcare, Chalfont, UK) in TBS-T for 1 h.
Finally, the membranes were incubated with Amersham ECL Western
Blotting Detection Reagent (GE Healthcare) and exposed to
BioMax® X-ray films (Kodak, Rochester, NY, USA) in a
dark room.
MTT-based cytotoxicity assay
In total, 100,000 cells were seeded in 35×10 mm
Corning plates. In the logarithmic phase of growth, the cells were
treated with various doses of BAPTA-AM and bortezomib (0.5 µM
BAPTA-AM, 5 µM BAPTA-AM, 1 nM bortezomib, 10 nM bortezomib, 0.5 µM
BAPTA-AM + 1 nM bortezomib, 0.5 µM BAPTA-AM + 10 nM bortezomib, 5
µM BAPTA-AM + 1 nM bortezomib and 5 µM BAPTA-AM + 10 nM bortezomib)
for 24 h. Following inhibitor exposure, the cells were treated for
24 h with RPMI-1640 media containing 0.5% FBS + 0.5 mg/ml MTT at
37°C with 5% CO2. Subsequently, the cells were incubated
with 3% SDS (200 µl) + 40 mM HCl/isopropanol (1 ml) for 15 min in
order to dissolve the MTT-formazan crystals. The absorbance of each
sample was recorded at 570 nm (13–15). Cell
survival was determined by analyzing the data with GraphPad Prism
3.03 software (GraphPad Software, Inc., La Jolla, CA, USA).
Half maximal inhibitory concentration
(IC50) determination
MTT assay was performed as previously described to
determine the IC50 of BAPTA-AM on 4T1 cells. The cells
were treated with 100 and 500 nM and 1, 10, 50 and 100 µM BAPTA-AM.
The IC50 value of BAPTA-AM was obtained by fitting the
data with a GraphPad Prism 3.03 program to a sigmoidal
dose-response curve.
WST-1-based cytotoxicity assay
In total, 1,000 cells were seeded in each well of a
96-well plate. In the logarithmic phase of growth, the cells were
treated with various doses of BAPTA-AM and bortezomib (5 µM
BAPTA-AM, 1 nM bortezomib, 10 nM bortezomib, 5 µM BAPTA-AM + 1 nM
bortezomib and 5 µM BAPTA-AM + 10 nM bortezomib) for 24 h.
Following inhibitor exposure, the cells were treated for 1 h with
RPMI-1640 media containing 0.5% FBS + 10 mg/ml WST-1 at 37°C with
5% CO2. Subsequently, the absorbance of the wells was
recorded by a microplate reader at 450 nm with a reference
wavelength at 630 nm.
iCELLigence system
In total, 12,500 cells were seeded onto an E-Plate
L8 within an iCELLigence system (ACEA Biosciences, San Diego, CA,
USA), which has integrated microelectrode sensors in the bottom of
the wells, and were incubated at 37°C with 5% CO2 for 96
h. In an iCELLigence system, as the cells proliferate they adhere
to the micro-electrodes, and alterations in electrical impedance
reflect the biological status of the cells; therefore, the system
allows monitoring of time-dependent effects on a cell culture. Cell
status is expressed as cell index (CI). In total 24 h later, in the
logarithmic phase of the growth, the cells were treated with 10 nM
bortezomib, 1 µM BAPTA-AM, 5 µM BAPTA-AM, 1 µM BAPTA-AM + 10 nM
bortezomib or 5 µM BAPTA-AM + 10 nM bortezomib. Following a 1 h
treatment time, CI measurements were taken.
Apoptotic DNA isolation
In total, 200,000 4T1 cells were seeded in 60×15 mm
sterile petri dishes and treated with 10 nM bortezomib, 1 µM
BAPTA-AM, 5 µM BAPTA-AM, 10 nM bortezomib + 1 µM BAPTA-AM or 10 nM
bortezomib + 5 µM BAPTA-AM at the logarithmic phase of growth for
24 h. Control cells were treated with dimethyl sulfoxide (vehicle
for BAPTA-AM). Following treatment, the cells were washed with 1 ml
phosphate-buffered saline (PBS) and resuspended in 200 µl PBS. An
Apoptotic DNA-Ladder kit (Roche Diagnostics GmbH) was used to
isolate DNA, according to the manufacturer's protocol. Equal
amounts of DNA (1 µg) from each sample were separated by 1.5%
agarose gel electrophoresis at 80 V for 2 h. DNA was visualized by
ethidium bromide staining under UV light.
Annexin V and dead cell analyses
In total, 200,000 4T1 cells were seeded in 60×15 mm
sterile petri dishes, and cells in the logarithmic phase of growth
were treated for 24 h with 10 nM bortezomib, 1 µM BAPTA-AM, 5 µM
BAPTA-AM, 10 nM bortezomib + 1 µM BAPTA-AM or 10 nM bortezomib + 5
µM BAPTA-AM. Following treatment, a 100 µl cell sample was prepared
using 40,000 cells and 1% FBS. In total, 100 µl Muse®
Annexin V and Dead Cell Reagent (EMD Millipore, Billerica, MA, USA)
was added to each sample. The cell samples were mixed thoroughly by
pipetting up and down or vortexing at a medium speed for 3–5 sec
and were left to stain for 20 min at room temperature in the dark.
Following staining, the apoptotic effects of the inhibitors were
analyzed by a Muse® Cell Analyzer (EMD Millipore).
Analysis of intracellular signaling
molecules
PathScan® Intracellular Signaling Array
kit (Cell Signaling Technology, Inc., Danvers, MA, USA) was used,
according to the manufacturer's protocol, for the detection of
phosphorylation or cleavage of the following 18 well-characterized
signaling molecules: Extracellular signal-regulated kinases 1/2
(Thr202/Tyr204); Signal transducer and activator of transcription
(Stat) 1 (Tyr701); Stat3 (Tyr705); Akt (Thr308); Akt (Ser473);
AMP-activated protein kinase α (Thr172); S6 ribosomal protein
(Ser235/236); mechanistic target of rapamycin (Ser2,448); Hsp27
(Ser78); Bcl-2-associated death promoter (Ser112); p70 S6 kinase
(Thr389); proline-rich Akt substrate of 40 kDa (Thr246); p53
(Ser15); p38 (Thr180/Tyr182); stress-activated protein and Jun
amino-terminal kinases (SAPK/JNK; Thr183/Tyr185); poly (ADP-ribose)
polymerase (Asp214); caspase-3 (Asp175); and glycogen synthase
kinase-3β (Ser9).
Briefly, 200,000 cells were seeded in 60×15 mm
sterile petri dishes and treated with 10 nM bortezomib, 100 nM
bortezomib, 1 µM BAPTA-AM, 5 µM BAPTA-AM, 10 nM bortezomib + 1 µM
BAPTA-AM, 10 nM bortezomib + 5 µM BAPTA-AM, 100 nM bortezomib + 1
µM BAPTA-AM or 100 nM bortezomib + 5 µM BAPTA-AM at the logarithmic
phase of the growth for 24 h. Following treatment, the medium was
removed and the cells were washed with ice-cold 1X PBS.
Subsequently, 0.3 ml ice-cold cell lysis buffer was added to each
plate and incubated on ice for 5 min. The lysate was centrifuged at
10,000 × g for 10 min at 4°C. The protein concentration was
determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) with bovine serum albumin as a standard.
Briefly, the glass slide array containing the antibodies for the
proteins to be analyzed was affixed to a multi-well plate.
Subsequently, 100 µl array blocking buffer was added to each well,
the wells were covered with sealing tape and incubated for 15 min
at room temperature on an orbital shaker. Following blocking, 75 µl
diluted lysate (45 µg protein) was added to each well and was
incubated for 2 h at room temperature on an orbital shaker.
Following washing, 75 µl 1X detection antibody cocktail was added
to each well and incubated for 1 h. Subsequently, 75 µl 1X
HRP-linked streptavidin was added to each well for 30 min at room
temperature. Following washing, the plate was incubated with
Lumi-GLO®/peroxide Reagent (Cell Signalling Technology,
Inc.) for 2 min and exposed to Kodak BioMax X-ray films in a dark
room.
Statistical analysis
Results were analyzed with GraphPad Prism 3.03
software. Statistical differences between the samples were
evaluated using one-way analysis of variance and Bonferroni or
Newman-Keuls post-hoc comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of bortezomib on GRP78
expression and proteasome inhibition
The clinical efficacy of proteasome inhibitor
bortezomib is hampered by drug-resistant cell phenotypes (16). Although there may be a number of
resistance mechanisms to bortezomib, one primary mechanism is the
constitutively high expression of Hsp27 (17). Initially, the present study
investigated the effect of bortezomib on GRP78 expression in a
dose- and time-dependent manner in metastatic mouse breast cancer
4T1 cells. The cells were treated with various doses of bortezomib
(10, 50, 100 and 200 nM) and 10 µM leupeptin, an inhibitor of
lysosomal proteases for 24 h, and GRP78 expression was determined
by western blot analysis. As shown in Fig. 1A, bortezomib treatment increased the
expression of GRP78 in a threshold-dependent manner. The highest
increase in GRP78 expression was observed in response to 10 nM
bortezomib treatment. With higher doses of bortezomib, there was a
slight decrease in GRP78 expression compared with 10 nM bortezomib.
In contrast to expectations, leupeptin also enhanced the expression
of GRP78. These results suggest that, in addition to the
proteasome, the lysosome plays a role in the regulation of GRP78
expression. To determine whether the proteasome was inhibited with
bortezomib treatment, the accumulation of polyubiquitin conjugates
were investigated by western blot analysis. As shown by Fig. 1A, polyubiquitin conjugates accumulated
in a dose-dependent manner, which indicated that the proteasome was
inhibited by bortezomib treatment.
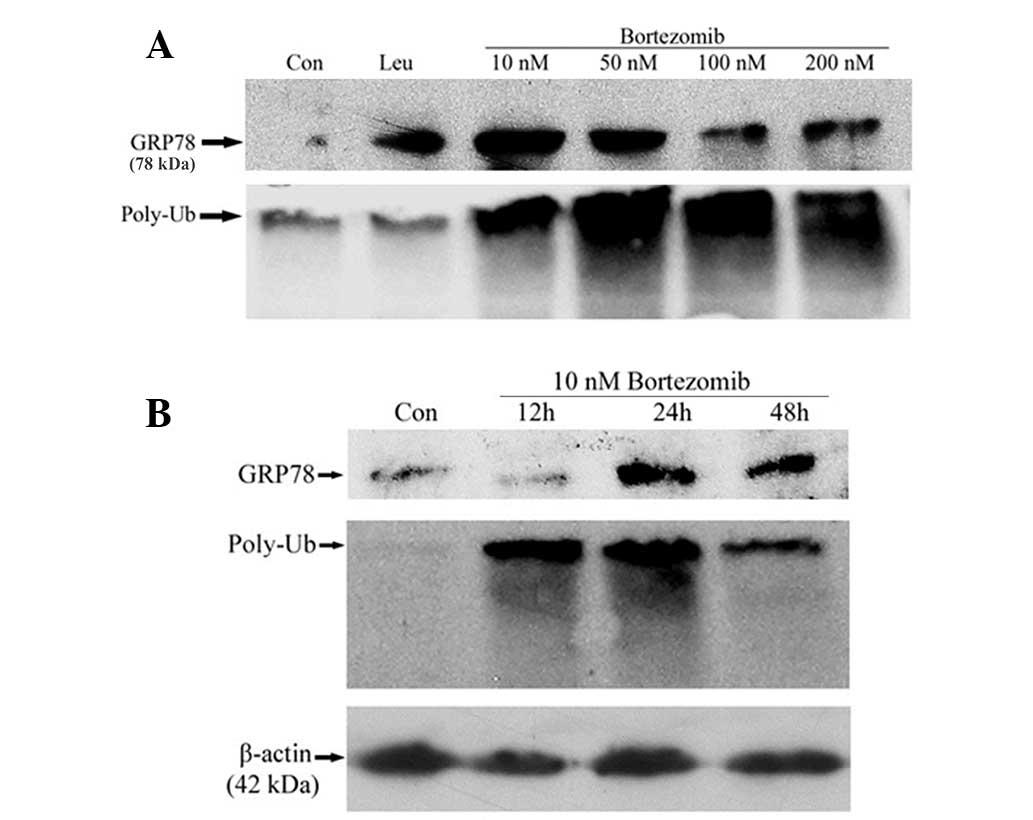 | Figure 1.(A) Expression of GRP78 in response to
various doses of bortezomib. Metastatic mouse breast cancer 4T1
cells were treated with bortezomib (10, 50, 50, 100 or 200 nM) and
10 µM leupeptin for 24 h. In total, 40 µg protein were separated by
12% SDS-PAGE and probed with GRP78 (1:500) and ubiquitin (1:200)
antibodies. Ubiquitin antibodies were used to determine the
accumulation of poly-Ub. (B) Time-dependent analysis of GRP78
following treatment with 10 nM bortezomib. The cells were treated
with bortezomib for 0, 12, 24 and 48 h. In total, 25 µg protein
were separated by 12% SDS-PAGE and probed with GRP78 (1:500) or
ubiquitin (1:200) antibodies. Equal protein loading was assessed
with a β-actin antibody. GRP78, glucose-regulated protein 78 kDa;
Con, control; Leu, leupeptin; poly-Ub, polyubiquitinated
conjugates; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel
electrophoresis. |
To determine the time-dependent effects of
bortezomib treatment, 4T1 cells were treated with 10 nM bortezomib
for 12, 24 and 48 h. Following 10 nM bortezomib treatment, there
was an increase in GRP78 expression at 24 h, which was sustained
until 48 h (Fig. 1B). By contrast, an
accumulation of polyubiquitin conjugates was observed as early as
12 h following 10 nM bortezomib-treatment (Fig. 1B), indicating that the increase in
GRP78 expression occurs following inhibition of the proteasome.
β-actin was used as a loading control.
Effects of the intracellular calcium
chelator BAPTA-AM
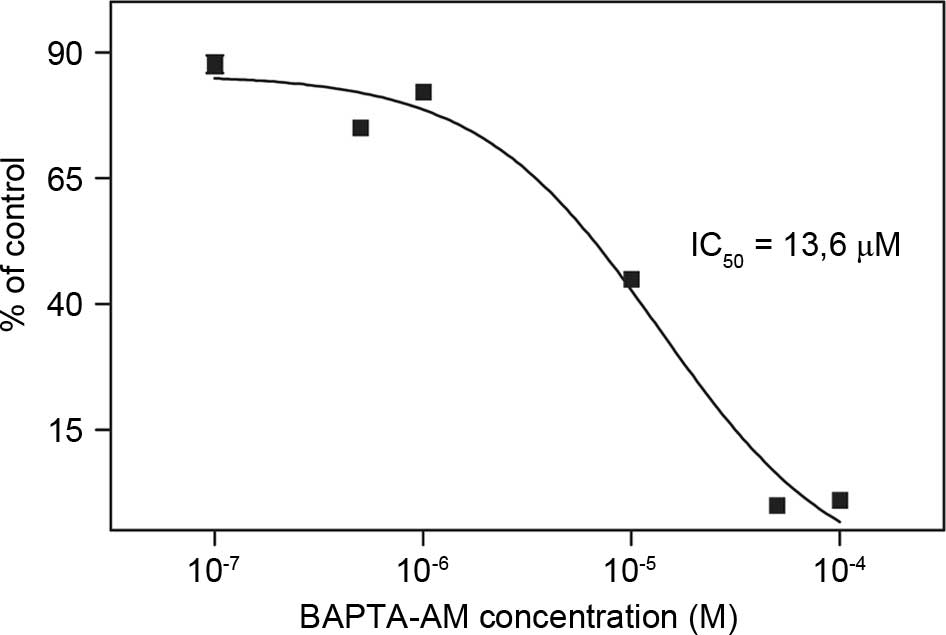
In order to determine the combined effects of
bortezomib and BAPTA-AM, the IC50 value of BAPTA-AM was
investigated using a MTT assay. The IC50 value of
BAPTA-AM was determined as 13.6 µM in the breast cancer 4T1 cell
line (Fig. 2). The IC50
value of bortezomib was previously determined as 71 nM in 4T1 cells
(15). Since a previous study showed
that the expression of antiapototic GRP78 protein may be suppressed
in the presence of BAPTA-AM (12),
the present study hypothesized that bortezomib + BAPTA-AM
combination may be more cytotoxic. Subsequently, the combined
effects of bortezomib and BAPTA-AM on the 4T1 cells was
investigated based on the IC50 value of BAPTA-AM
determined by the present study. The cells were treated with
various doses of bortezomib (1 nM and 10 nM) and BAPTA-AM (0.5 and
5 µM). As shown in Fig. 3A, the
combination of 10 nM bortezomib + 5 µM BAPTA-AM was more effective
compared with monotherapies (10 nM bortezomib or 5 µM BAPTA-AM
alone) (P<0.05). A WST-1 assay was used to verify the findings
obtained by MTT assay and similar results were obtained; the
combination of 10 nM bortezomib + 5 µM BAPTA was again
significantly different compared with 10 nM bortezomib or 5 µM
BAPTA-AM treatment alone (P<0.001; Fig. 3B).
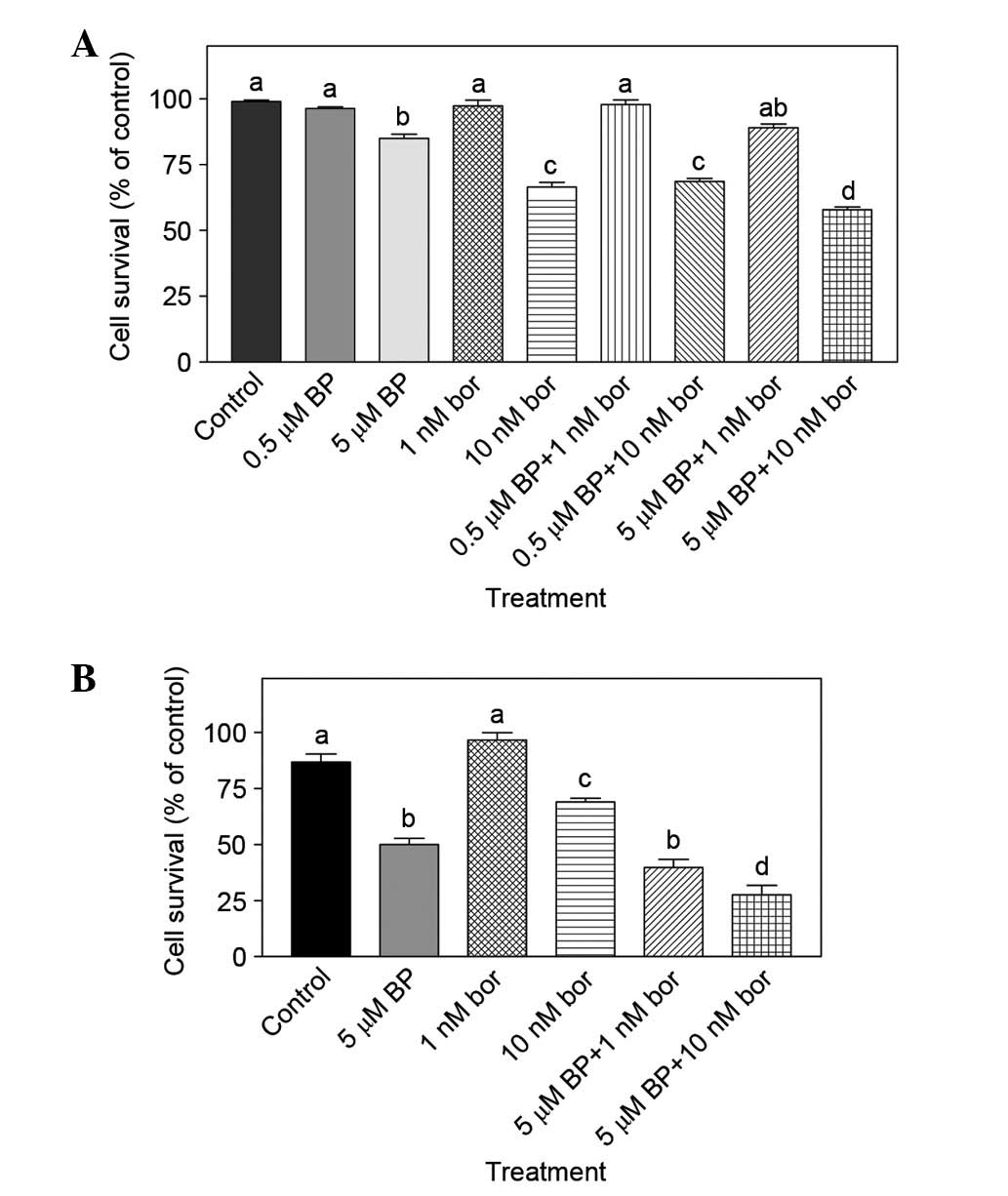 | Figure 3.(A) Combined effect of bor and BP
determined by MTT assay. Mouse breast cancer 4T1 cells were treated
with 0.5 µM BP, 5 µM BP, 1 nM bor, 10 nM bor alone or 0.5 µM BP + 1
nM bor 0.5 µM BP + 10 nM bor, 5 µM BP + 1 nM bor or 5 µM BP + 10 nM
bor for 24 h. The number of viable cells was determined by MTT
assay (n=3). (B) Combined effect of bor and BP determined by WST-1
assay. Cells were treated with 5 µM BP, 1 nM bor or 10 nM bor alone
or 5 µM BP + 1 nM bor and 5 µM BP + 10 nM bor for 24 h. The number
of viable cells was determined by WST-1 assay (n=8–9). Results are
presented as the mean ± standard error of the mean. Mean values
that do not share a letter in common are significantly different
(P<0.05). BP, BAPTA-AM; bor, bortezomib; MTT,
3-(4,5-dimethylthylthiazol-2-yl)-2,5-diphenyl-tetrazolium
bromide. |
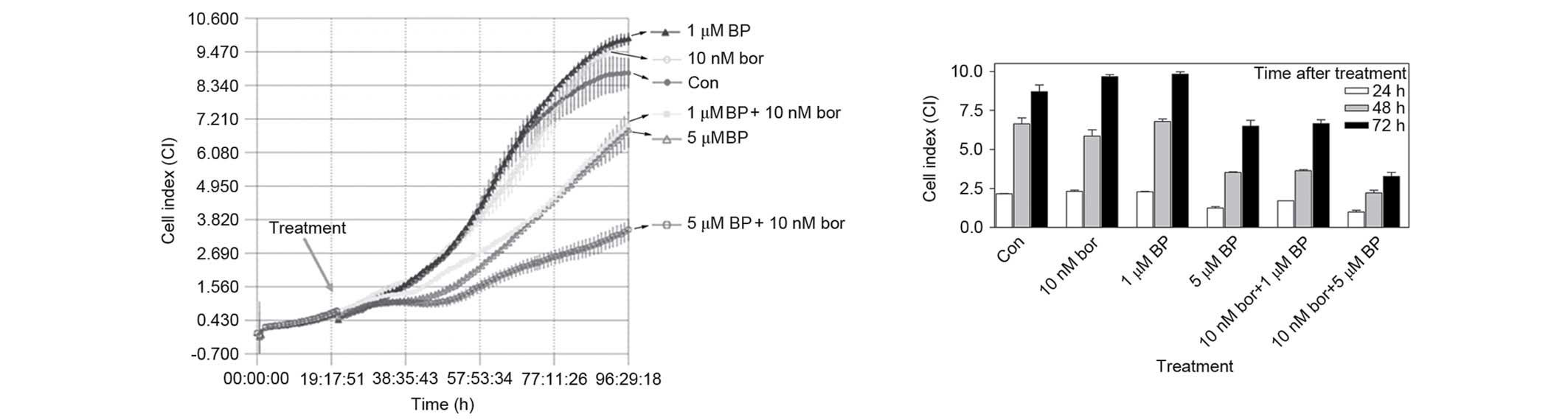
Effect of bortezomib + BAPTA-AM
treatment on various cell processes
The combined effect of various concentrations of
bortezomib + BAPTA-AM were investigated by the iCELLigence system,
which is an impedance-based system for real-time monitoring of
cellular processes, including cell growth, proliferation or
cytotoxicity. In total, 12,500 cells, seeded onto E-Plates L8, were
treated with 10 nM bortezomib, 1 µM BAPTA-AM, 5 µM BAPTA-AM and the
combinations of 10 nM bortezomib + 1 µM BAPTA-AM or 10 nM
bortezomib + 5 µM BAPTA-AM in the logarithmic phase of growth. As
shown by Fig. 4, although cells
treated with 1 µM BAPTA-AM and 10 nM bortezomib were not different
from the control, the combination of 10 nM bortezomib + 1 µM
BAPTA-AM was significantly different compared with monotherapy
following 24 h (P<0.01), 48 h (P<0.001) or 72 h (P<0.01)
of treatment. Similarly, 10 nM bortezomib + 5 µM BAPTA-AM reduced
the growth of cells significantly compared with 10 nM bortezomib
treatment following 24 h (P<0.001), 48 h (P<0.001) or 72 h
(P<0.001) of treatment. The same combinations also produced
significant results compared with 5 µM BAPTA-AM alone following 24
h (P<0.05), 48 h (P<0.05) or 72 h (P<0.001) treatment
(Fig. 4).
Effects of bortezomib + BAPTA-AM on
cell death
To determine whether cell death was mediated by
necrosis or apoptosis in response to treatment with bortezomib +
BAPTA-AM, DNA fragmentation was examined using a Roche Apoptotic
DNA-ladder kit. As shown in Fig. 5A,
DNA was not cleaved in response to a low dose of bortezomib (10
nM), 1 µM BAPTA-AM, 5 µM BAPTA-AM or 10 nM bortezomib + 1 µM
BAPTA-AM after 24 h; however, a clear increase in DNA fragmentation
(as determined by smearing) was observed following treatment with
10 nM bortezomib + 5 µM BAPTA-AM for 24 h (Fig. 5A). To confirm that the combination of
bortezomib + BAPTA-AM causes cell death primarily through
apoptosis, the effect of each inhibitor alone or as combination in
4T1 cells was analyzed using Muse® Annexin V and Dead
Cell Reagent. In control cells and cells treated with 10 nM
bortezomib, 1 µM BAPTA-AM and 5 µM BAPTA-AM, 6.3, 6.85, 6.05 and
9.65% early apoptotic cells were detected, respectively, following
24 h of treatment (Fig. 5B). By
contrast, 10.8 and 15.35% of cells were early apoptotic following
24 h treatment with 10 nM bortezomib + 1 µM BAPTA-AM and 10 nM
bortezomib + 5 µM BAPTA-AM combinations, respectively. This
suggests that 10 nM bortezomib + 5 µM BAPTA-AM combination is more
effective compared with inhibitor treatment alone or 10 nM
bortezomib + 1 µM BAPTA-AM combination (Fig. 5B).
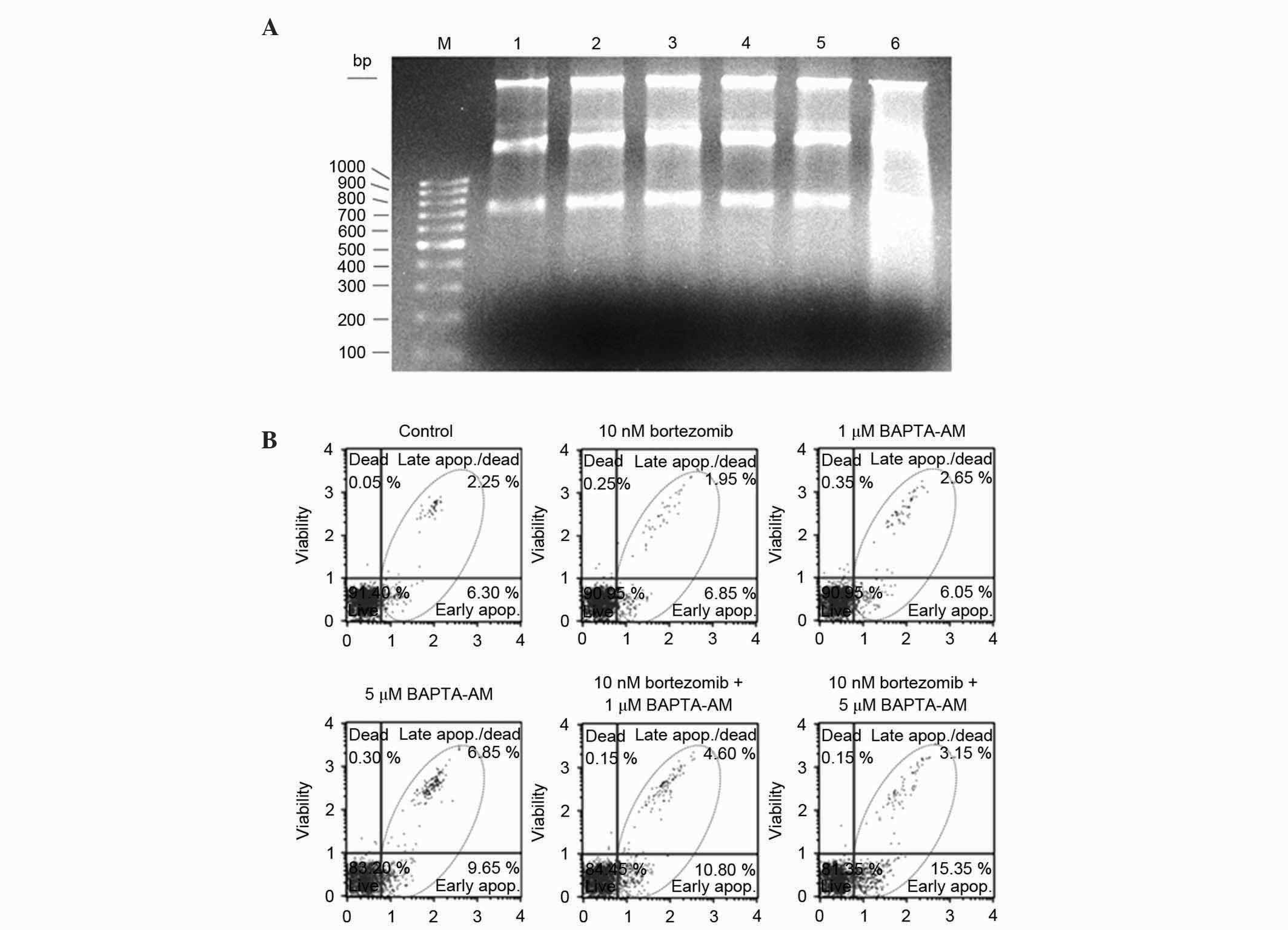 | Figure 5.(A) Analyses of DNA fragmentation by
Roche Apoptotic DNA-Ladder kit. Metastatic mouse breast cancer 4T1
cells were treated for 24 h with 10 nM bortezomib, 1 µM BAPTA-AM, 5
µM BAPTA-AM, 10 nM bortezomib + 1 µM BAPTA-AM and 10 nM bortezomib
+ 5 µM BAPTA-AM at the logarithmic phase of growth. Following
treatment, DNA was isolated using the Roche Apoptotic DNA Ladder
kit. For each DNA sample, equal amounts of DNA (1 µg) was separated
on a 1.5% agarose gel. M, 100 bp marker; lane 1, control; lane 2,
10 nM bortezomib; lane 3, 1 µM BAPTA-AM; lane 4, 5 µM BAPTA-AM;
lane 5, 10 nM bortezomib + 1 µM BAPTA-AM; lane 6, 10 nM bortezomib
+ 5 µM BAPTA-AM. (B) Determination of apoptotic effects of
bortezomib and BAPTA-AM by MUSE® Cell Analyzer. 4T1
cells were treated in the logarithmic phase of growth with 10 nM
bortezomib, 1 µM BAPTA-AM, 5 µM BAPTA-AM, 10 nM bortezomib + 1 µM
BAPTA-AM or 10 nM bortezomib + 5 µM BAPTA-AM for 24 h. Following
treatment, the cells from each sample were stained using Annexin V
and Dead Cell Reagent and analyzed by MUSE® Cell
Analyzer. Results are representative of at least two
experiments. |
Determination of the bortezomib +
BAPTA-AM mechanism
To determine the growth-inhibitory and apoptotic
mechanisms of bortezomib + BAPTA-AM combination, the
activation/inactivation of various proteins and enzymes were
analyzed by PathScan® Intracellular Signalling Array
kit, which is a slide-based antibody array that uses sandwich
immunoassay to detect phosphorylation or cleavage of 18 important
signalling molecules. As shown by Fig.
6, although 10 nM bortezomib, 1 µM BAPTA-AM, 5 µM BAPTA-AM and
10 nM bortezomib + 1 µM BAPTA-AM did not affect the phosphorylation
level of SAPK/JNK, there was an 86% increase in SAPK/JNK
phosphorylation with 10 nM bortezomib + 5 µM BAPTA-AM treatment,
which suggests that the growth-inhibitory and apoptotic mechanisms
may be mediated through the activation of SAPK/JNK. In addition, a
significant increase in SAPK/JNK phosphorylation was detected in
response to 100 nM bortezomib alone, 100 nM bortezomib + 1 µM
BAPTA-AM and 100 nM bortezomib + 5 µM BAPTA-AM (Fig. 6). There were 3.2, 3.7 and 3.9 fold
increases observed following treatments with 100 nM bortezomib, 100
nM bortezomib + 1 µM BAPTA-AM and 100 nM bortezomib + 5 µM
BAPTA-AM, treatment, respectively as compared to the DMSO-treated
control. As shown in Fig. 6, analysis
of the PathScan results indicated that there is no alterations in
the phosphorylation or cleavage of the other 17 signalling
molecules under the experimental conditions. This result may be
partly due to the fact that the antibodies may not have recognized
the mouse proteins, since 4T1 breast cancer cells are derived from
BALB/c mice.
 | Figure 6.PathScan® Intracellular
Array analysis. Metastatic mouse breast cancer 4T1 cells were
treated with 10 nM bor, 100 nM bor, 1 µM BAPTA-AM, 5 µM BP alone or
10 nM bor + 1 µM BP, 10 nM bor + 5 µM BP, 100 nM bor + 1 µM BP or
100 nM bor + 5 µM BP at the logarithmic phase of growth for 24 h.
Following treatment, the cells were analyzed using the
PathScan® Intracellular Signalling Array kit, according
to the manufacturer's protocol. The slide was incubated with
Lumi-GLO/peroxide reagent and exposed to Kodak BioMax X-ray films
in dark room. The result is representative of two experiments, each
run in duplicate. BP, BAPTA-AM; bor, bortezomib; SAPK/JNK,
stress-activated protein and Jun amino-terminal kinases. |
Discussion
GRP78 overexpression has been observed in a number
of malignant cells due to stress-inducing factors, including
nutrient deprivation, hypoxia and acidosis in the microenvironment
of poorly-perfused solid tumors (18). The induction of GRP78 favors cancer
cell survival and also confers drug resistant phenotypes (6). Therefore, the present study investigated
the expression of GRP78 following treatment with various doses of
bortezomib in the metastatic mouse breast cancer 4T1 cell line,
which is a p53-null cell line (15) that is commonly used as an in
vivo tumor formation model. The present results revealed that
the expression level of GRP78 was increased significantly in a
threshold- and time-dependent manner following low doses of
bortezomib. In addition, the results suggest for the first time, to
the best of our knowledge, that the 26S proteasome and lysosomal
organelle are involved in the regulation of GRP78 protein. Since
GRP78 is an antiapoptotic protein that has been revealed to be
responsible for chemotherapeutic resistance in a number of tumors
and cell lines (6,7), the present study rationalized that the
therapeutic response to bortezomib may be increased by simultaneous
inhibition of GRP78. Experiments performed in the present study
revealed that a combination of a low dose of bortezomib (10 nM)
with 5 µM BAPTA-AM, a strong inhibitor of GRP78 (19), caused significant cytotoxicity
compared with monotherapy of bortezomib and BAPTA-AM, as determined
by MTT assay. To verify these results, the cells were similarly
treated with the same combination of drugs and cell viability was
determined by WST-1 assay, which produced similar results as the
MTT assay. An iCELLigence system, which allows real-time monitoring
of cellular processes and offers distinct and important advantages
over traditional end-point assays, revealed that 10 nM bortezomib +
1 µM BAPTA-AM and 10 nM bortezomib + 5 µM BAPTA-AM combination
therapies reduced cell proliferation significantly compared with
monotherapies for up to 76 h of treatment. These results not only
demonstrate that combination therapy is more effective than
monotherapy, but also indicate that the inhibitors are either
stable for up to 72 h of incubation or damage the cells so that
cell proliferation is clearly decreased at longer incubation
times.
In addition, the present results revealed that 10 nM
bortezomib + 5 µM BAPTA-AM combination treatment causes
growth-inhibition and cell death through the induction of
programmed cell death (apoptosis). To determine the mechanism of
growth-inhibition and apoptosis, the phosphorylation or cleavage of
18 well-known signalling molecules was examined by PathScan
analysis. This analysis revealed that there was a significant
increase in the phosphorylation of SAPK/JNK following treatment
with 100 nM bortezomib alone or a combination of 10 nM bortezomib +
5 µM BAPTA-AM, 100 nM bortezomib + 1 µM BAPTA-AM or 100 nM
bortezomib + 5 µM BAPTA-AM. SAPK/JNK belongs to the
mitogen-activated protein kinase family, which is involved in the
regulation of cell proliferation, differentiation and apoptosis
(20). SAPK/JNK was originally
identified as a stress-activated kinase linked to the cell death
response, and is known to be activated by several stimuli,
including growth factors, cytokines and stress factors (20). Proteasomal inhibition by MG132 was
previously observed to potentiate leukemic cell apoptosis induced
by flavopiridol through a SAPK/JNK- and NF-κB-dependent process
(21). Similarly, exposure of chronic
myeloid leukemia K562 and LAMA84 cells to bortezomib alone resulted
in increased phosphorylation of SAPK/JNK (22). However, the findings presented by the
current study revealed for the first time, to the best of our
knowledge, that bortezomib alone or bortezomib + BAPTA-AM induced
apoptosis in breast cancer 4T1 cells, which is associated with the
phosphorylation/activation of SAPK/JNK.
A previous study revealed that there was an
association between loss of sensitivity to the proteasome inhibitor
and upregulation of the prosurvival chaperone GRP78 in mantle cell
lymphoma samples and cell lines (23). In addition, Kern et al
(24) indicated that
bortezomib-resistant solid tumor PC-3 and HRT-18 cell lines were
capable of secreting high amounts of GRP78. Kardosh et al
(25) demonstrated that small
interfering RNA-mediated knockdown of GRP78 renders tumor cells
more sensitive to a combination of bortezomib and celecoxib
treatment.
The present authors are currently investigating
whether the expression of GRP78 is downregulated by BAPTA-AM in
response to proteasome inhibitor bortezomib in 4T1 breast cancer
cells. Overall, the results from the present study suggest that
treatment protocols involving proteasome inhibitors in combination
with BAPTA-AM may produce more effective and beneficial outcomes in
the therapeutic responses of cancer patients harboring either a
wild-type or mutant p53 gene. The present study also hypothesizes
that the chemotherapy resistance in cancer cells may be prevented
by simultaneous treatment of cancer cells with the proteasome
inhibitor bortezomib and BAPTA-AM.
Acknowledgements
The present study was financially supported by the
Scientific and Technological Research Council of Turkey (project
no., 113S400).
References
|
1
|
Hendershot LM: The ER function BiP is a
master regulator of ER function. Mt Sinai J Med. 71:289–297.
2004.PubMed/NCBI
|
|
2
|
Hendershot LM, Valentine VA, Lee AS,
Morris SW and Shapiro DN: Localization of the gene encoding human
BiP/GRP78, the endoplasmic reticulum cognate of the HSP70 family,
to chromosome 9q34. Genomics. 20:281–284. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Murphy ME: The HSP70 family and cancer.
Carcinogenesis. 34:1181–1188. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roller C and Maddalo D: The molecular
chaperone GRP78/BiP in the development of chemoresistance:
Mechanism and possible treatment. Front Pharmacol. 4:102013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Qiu W, Kohen-Avramoglu R, Mhapsekar S,
Tsai J, Austin RC and Adeli K: Glucosamine-induced endoplasmic
reticulum stress promotes ApoB100 degradation: Evidence for
Grp78-mediated targeting to proteasomal degradation. Arterioscler
Thromb Vasc Biol. 25:571–577. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R
and Lee AS: Cancer cells resistant to therapy promote cell surface
relocalization of GRP78 which complexes with PI3K and enhances
PI(3,4,5)P3 production. PLoS One. 8:e800712013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li B, Cheng XL, Yang YP and Li ZQ: GRP78
mediates radiation resistance of a stem cell-like subpopulation
within the MCF-7 breast cancer cell line. Oncol Rep. 30:2119–2126.
2013.PubMed/NCBI
|
|
8
|
Dong D, Ko B, Baumeister P, Swenson S,
Costa F, Markland F, Stiles C, Patterson JB, Bates SE and Lee AS:
Vascular targeting and antiangiogenesis agents induce drug
resistance effector GRP78 within the tumor microenvironment. Cancer
Res. 65:5785–5791. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J, Jiang Y, Jia Z, Li Q, Gong W,
Wang L, Wei D, Yao J, Fang S and Xie K: Association of elevated
GRP78 expression with increased lymph node metastasis and poor
prognosis in patients with gastric cancer. Clin Exp Metastasis.
23:401–410. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang HQ, Du ZX, Zhang HY and Gao DX:
Different induction of GRP78 and CHOP as a predictor of sensitivity
to proteasome inhibitors in thyroid cancer cells. Endocrinology.
148:3258–3270. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mozos A, Roué G, López-Guillermo A, Jares
P, Campo E, Colomer D and Martinez A: The expression of the
endoplasmic reticulum stress sensor BiP/GRP78 predicts response to
chemotherapy and determines the efficacy of proteasome inhibitors
in diffuse large b-cell lymphoma. Am J Pathol. 179:2601–2610. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen LY, Chiang AS, Hung JJ, Hung HI and
Lai YK: Thapsigargin-induced grp78 expression is mediated by the
increase of cytosolic free calcium in 9l rat brain tumor cells. J
Cell Biochem. 78:404–416. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Freshney RI: Culture of Animal Cells: A
Manual of Basic Technique. Wiley-Liss. Hoboken, NJ: 2005.
View Article : Google Scholar
|
|
14
|
Savran B, Yerlikaya A, Erdoğan E and Genç
O: Anticancer agent ukrain and bortezomib combination is
synergistic in 4T1 breast cancer cells. Anticancer Agents Med Chem.
14:466–472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yerlikaya A and Erin N: Differential
sensitivity of breast cancer and melanoma cells to proteasome
inhibitor velcade. Int J Mol Med. 22:817–823. 2008.PubMed/NCBI
|
|
16
|
Oerlemans R, Franke NE, Assaraf YG, Cloos
J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova
K, Lemos C, et al: Molecular basis of bortezomib resistance:
Proteasome subunit beta5 (PSMB5) gene mutation and overexpression
of PSMB5 protein. Blood. 112:2489–2499. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chauhan D, Li G, Shringarpure R, Podar K,
Ohtake Y, Hideshima T and Anderson KC: Blockade of Hsp27 overcomes
bortezomib/proteasome inhibitor PS-341 resistance in lymphoma
cells. Cancer Res. 63:6174–6177. 2003.PubMed/NCBI
|
|
18
|
Ni M, Zhang Y and Lee AS: Beyond the
endoplasmic reticulum: Atypical GRP78 in cell viability, signalling
and therapeutic targeting. Biochem J. 434:181–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Wang W, Wang S, Wang J, Shao S and
Wang Q: Down-regulation of GRP78 is associated with the sensitivity
of chemotherapy to VP-16 in small cell lung cancer NCI-H446 cells.
BMC Cancer. 8:3722008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dai Y, Rahmani M and Grant S: Proteasome
inhibitors potentiate leukemic cell apoptosis induced by the
cyclin-dependent kinase inhibitor flavopiridol through a SAPK/JNK-
and NF-kappaB-dependent process. Oncogene. 22:7108–7122. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dai Y, Rahmani M, Pei XY, Dent P and Grant
S: Bortezomib and flavopiridol interact synergistically to induce
apoptosis in chronic myeloid leukemia cells resistant to imatinib
mesylate through both Bcr/Abl-dependent and -independent
mechanisms. Blood. 104:509–518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Roué G, Pérez-Galan P, Mozos A,
López-Guerra M, Xargay-Torrent S, Rosich L, Saborit-Villarroya I,
Normant E, Campo E and Colomer D: The Hsp90 inhibitor IPI-504
overcomes bortezomib resistance in mantle cell lymphoma in vitro
and in vivo by down-regulation of the prosurvival ER chaperone
BiP/Grp78. Blood. 117:1270–1279. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kern J, Untergasser G, Zenzmaier C, Sarg
B, Gastl G, Gunsilius E and Steurer M: GRP-78 secreted by tumor
cells blocks the antiangiogenic activity of bortezomib. Blood.
114:3960–3967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kardosh A, Golden EB, Pyrko P, Uddin J,
Hofman FM, Chen TC, Louie SG, Petasis NA and Schönthal AH:
Aggravated endoplasmic reticulum stress as a basis for enhanced
glioblastoma cell killing by bortezomib in combination with
celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer
Res. 68:843–851. 2008. View Article : Google Scholar : PubMed/NCBI
|