Introduction
Familial adenomatous polyposis (FAP) is an autosomal
dominantly inherited disease (1),
which is characterized by the development of hundreds to thousands
of adenomatous polyps in the colon and rectum (2) during the second and third decades of
life. If these polyps are not removed, most FAP patients progress
to colorectal cancer (CRC) (3). It is
well known that FAP is linked to germline mutations of the
adenomatous polyposis coli (APC) gene (5q21-q22; OMIM #175100)
(4,5),
which consists of 15 coding exons and an 8538 bp open reading
frame. In 1991, Groden et al described FAP as an
autosomal-dominant disorder caused by germline mutations of APC
(5). The examination of APC germline
mutations in FAP families is currently considered an efficient tool
for predictive testing of subjects at risk. To date, over 1500
different pathogenic APC mutations have been reported in the Leiden
Open Variation Database (http://www.lovd.nl/2.0/).
At present, a number studies on the genetic
screening of FAP in China (6–9) and other countries have been reported;
however, the detection rate and the associated mutations vary
markedly (10–15). A number of patients with FAP have no
APC mutation. Among the known genes, evidence from previous studies
suggests that MUTYH mutations observed in FAP patients may explain
up to 25% of ‘APC mutation-negative’ FAP (16–18). In
addition, AXIN2, which is involved in the Wnt signaling pathway,
has also been selected as a candidate gene for FAP (19).
A large number of factors which might predispose
individuals to FAP have been reported; however, further analysis of
the pathogenesis of disease-associated variants still presents a
significant challenge and huge cost. With the rapid development of
science and technology, tools and databases for the prediction of
pathogenicity have been promoted, and the tests to verify the
predicted results are inspiring. For example, certain synonymous
mutations, which were sometimes considered as silent mutations, are
now widely acknowledged to be capable of causing changes in protein
expression, conformation and function.
In our study, in order to investigate germline
mutations in Chinese patients with FAP, a total of 11 FAP patients
were selected for exon-specific DNA sequencing of the APC gene to
determine the micromutation type. For samples without known
pathogenic changes predisposing to FAP in the APC gene, whole-gene
sequencing of the MUTYH gene and AXIN2 gene was further performed.
Due to the high cost and the long time required for the analysis of
huge amounts of data, the pathogenicity of amino acid substitution
identified in our study was tested and predicted by computational
methods.
Patients and methods
Ethics
This study was approved by and carried out in
accordance with the Research Ethics Board of the First Affiliated
Hospital of Kunming Medical University (Kunming, China). All
subjects provided their written informed consent to participate in
this study. The ethics committees approved the consent
procedure.
Subject recruitment
From 2005 to 2013, 11 FAP patients were recruited
from the Department of Oncology, at the First Affiliated Hospital
of Kunming Medical University. All patients were referred to this
study initially based on clinically suspicious colonoscopic
findings, and the diagnosis of FAP was subsequently confirmed by
surgery or endoscopic mucosal resection and through a suggestive
family history of FAP.
Genomic DNA purification
Peripheral venous blood (10 ml) was drawn from FAP
patients and genomic DNA was purified following the instructions of
the DNA extraction kit (QIAamp DNA blood mini kit, Qiagen,
Valencia, CA, USA). The extracted DNA was quantified by a Pearl
nanophotometer (Implen, Munich, Germany).
Exon-specific DNA sequencing of
APC
All FAP patients underwent exon-specific DNA
sequencing of APC. The 1–14 exons of the promoter region and the 21
segments of the 15th exon in the APC gene were amplified separately
by polymerase chain reaction (PCR). The primer pairs were designed
as previously described (20). All
samples were amplified in 20 µl reaction mixture containing 50 ng
genomic DNA, 10 mM Tris-HCl, 50 mM KCl, 2.5 mM MgCl2,
0.2 mM each dNTP, 200 mM each primer, 5% DMSO and 0.01 units Ex Taq
polymerase (Takara, Tokyo, Japan). The thermal cycling profile was
composed of an initial denaturation step at 94°C for 5 min, 30
cycles of 30 sec of denaturation at 94°C, 30 sec of annealing at
58°C, and 45 sec of extension at 72°C, with a final 10-min
extension step at 72°C. After observing by agarose gel
electrophoresis, PCR products were purified using a MiniBEST
Universal DNA extraction kit (Takara). DNA sequencing reactions
were performed using a BigDye Terminator v3.1 Cycle sequencing kit
(Applied Biosystems, Foster City, CA, USA). Nucleotide sequences of
both strands were determined with an ABI Prism 3730 genetic
analyzer (Applied Biosystems).
Whole-gene sequencing of MUTYH
For the six patients without direct pathogenic
changes predisposing to FAP in the APC gene (i.e., patients for
whom samples presented synonymous mutations only), MUTYH mutation
screening was carried out. Whole-gene sequencing of the MUTYH gene
was performed; the primer pairs used for MUTYH sequencing in this
study were designed as previously described (21). All samples were amplified in 20 µl
reaction mixture. The PCR cycling profile was composed of an
initial denaturation step at 95°C for 5 min, 30 cycles of 30 sec of
denaturation at 95°C, 1 min of annealing at 57°C, and 1.5 min of
extension at 72°C, with a final 8-min extension step at 72°C. After
purifying the PCR products, DNA sequencing was applied and
analyzed.
AXIN2 mutation screening
The Wnt signaling regulator gene AXIN2 was tested in
the same batch of samples following the screening of the MUTYH
gene. The coding region of AXIN2 was sequenced for mutations exon
by exon using primers and conditions as previously described
(19). The PCR products were analyzed
by DNA sequencing.
Mutation analysis
The sequencing data of each gene tested were firstly
analyzed using Mutation Surveyor by SoftGenetics (http://www.softgenetics.com/). For mutations of the
APC gene, the UMD APC mutations database (http://www.umd.be/APC/), APC-Database (http://www.LOVD.nl/APC), Zhejiang University-Adinovo
Center APC Database (22,23) (http://www.genomed.org/lovd2/home.php?select_db=APC)
and the APC Mutation Database (http://fap.taenzer.me) were applied to screen for the
presence of novel and already-known variants. For the mutation
screening of the MUTYH gene, the MUTYH Mutation Database
(http://www.LOVD.nl/MUTYH) was used. For mutations
of the AXIN2 gene, Zhejiang University Center for Genetic and
Genomic Medicine (ZJU-CGGM) (22,23)
(http://www.genomed.org/lovd2/home.php?select_db=AXIN2)
was consulted. In addition to the above-mentioned tools and
websites for the mutation screening of each gene, the Human Gene
Mutation Database (http://www.hgmd.org/), International HapMap Project
(www.hapmap.org/), Nucleotide Variation and
Mutation Database (http://www.mutationdiscovery.com/md/MD.com/home_page.jsp),
dbSNP database (24) (http://www.ncbi.nlm.nih.gov/SNP/), 1000 Genomes
(http://www.1000genomes.org/), and
Ensembl (25) (www.ensembl.org/) were also used.
In silico analysis
Mutations identified from each gene identified in
our study were analyzed using an extensive set of assessment tools
and databases. For the already known amino acid substitutions,
their corresponding SNP IDs were confirmed. For each mutation, a
series of tests was executed to determine whether the mutation had
a functional impact with respect to functional categories including
protein coding, splicing regulation, transcriptional regulation and
post-translation. For the prediction of deleterious missense
mutations, PolyPhen-2 (26), SIFT
(27) and SNPs&GO (28–30) were
used. For the illustration of conserved amino acids, Pfam
(http://pfam.sanger.ac.uk/) (31), Clustal W2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (32) and Jalview (http://www.jalview.org/) (33) were used. The functional consequences
of synonymous and intronic mutations identified in our study were
predicted by the splicing prediction programs ESEfinder (34), ESRSearch (35), PESX (36) and RESCUE_ESE (37).
Results
Patient characteristics
Eleven Chinese FAP patients with a clinical
diagnosis of classical were included in this study. The group
comprised 6 males and 5 females (mean age, 28 years; range, 18–36
years). The age and gender distribution between the two groups were
not significantly different. The patient characteristics are
summarized in Table I.
 | Table I.Characteristics of familial
adenomatous polyposis patients (n=11) treated at our institution
between 2005 and 2013. |
Table I.
Characteristics of familial
adenomatous polyposis patients (n=11) treated at our institution
between 2005 and 2013.
| Age | Gender | Number of
adenomas | Treatment | Diagnosis of
CRC |
|---|
| 29 | M | ﹥1,000 |
Surgerya | Yes |
| 22 | M | ﹥1,000 | Surgery | Yes |
| 35 | M | ﹥1,000 | Surgery | Yes |
| 36 | M | 500–1,000 | Surgery | Yes |
| 26 | M | ﹥1,000 | Surgery | Yes |
| 25 | M | ﹥1,000 | Surgery | Yes |
| 32 | F | ﹥1,000 | Surgery | Yes |
| 29 | F | ﹥1,000 | Surgery | Yes |
| 29 | F | 100–200 | EMR | No |
| 18 | F | ﹥1,000 | Surgery | Yes |
| 27 | F | ﹥1,000 | Surgery | Yes |
Micromutations of each gene
After DNA sequencing of the APC, MUTYH and AXIN2
genes, the micromutations of each gene were analyzed by Mutation
Surveyor, and the mutations are summarized in Table II. Eleven different types of APC
polymorphism were observed in the cohort of the families analyzed,
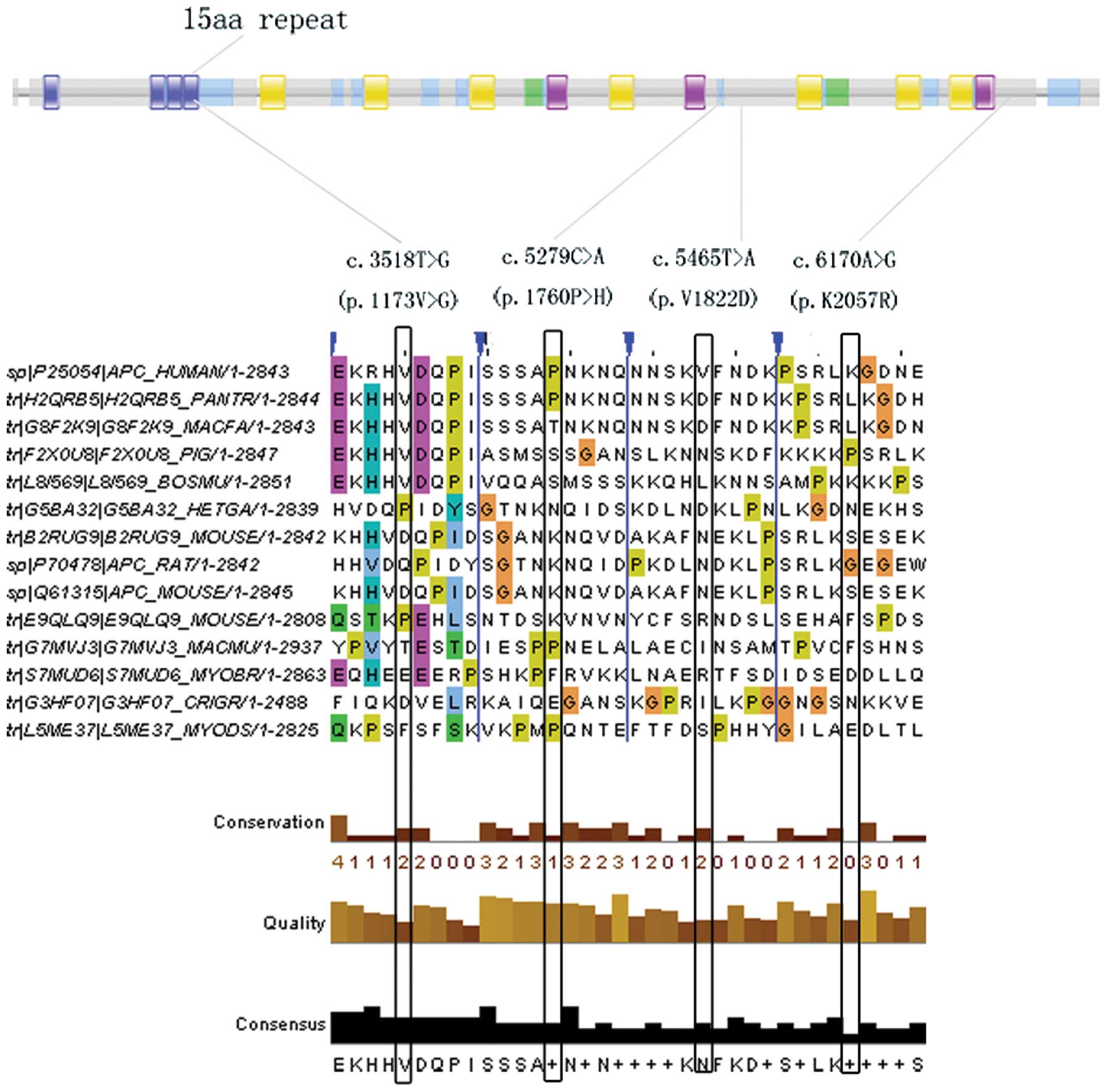
of which four were missense substitutions (V1822D, V1173G, P1760H
and K2057), six were silent substitutions (Y486Y, T449T, T1493T,
G1678G, S1756S and P1960P) and one was a nonsense substitution
(S1196X). The nonsense substitution (S1196X) in the APC gene was an
already-known pathogenic mutation, which resulted in a stop codon
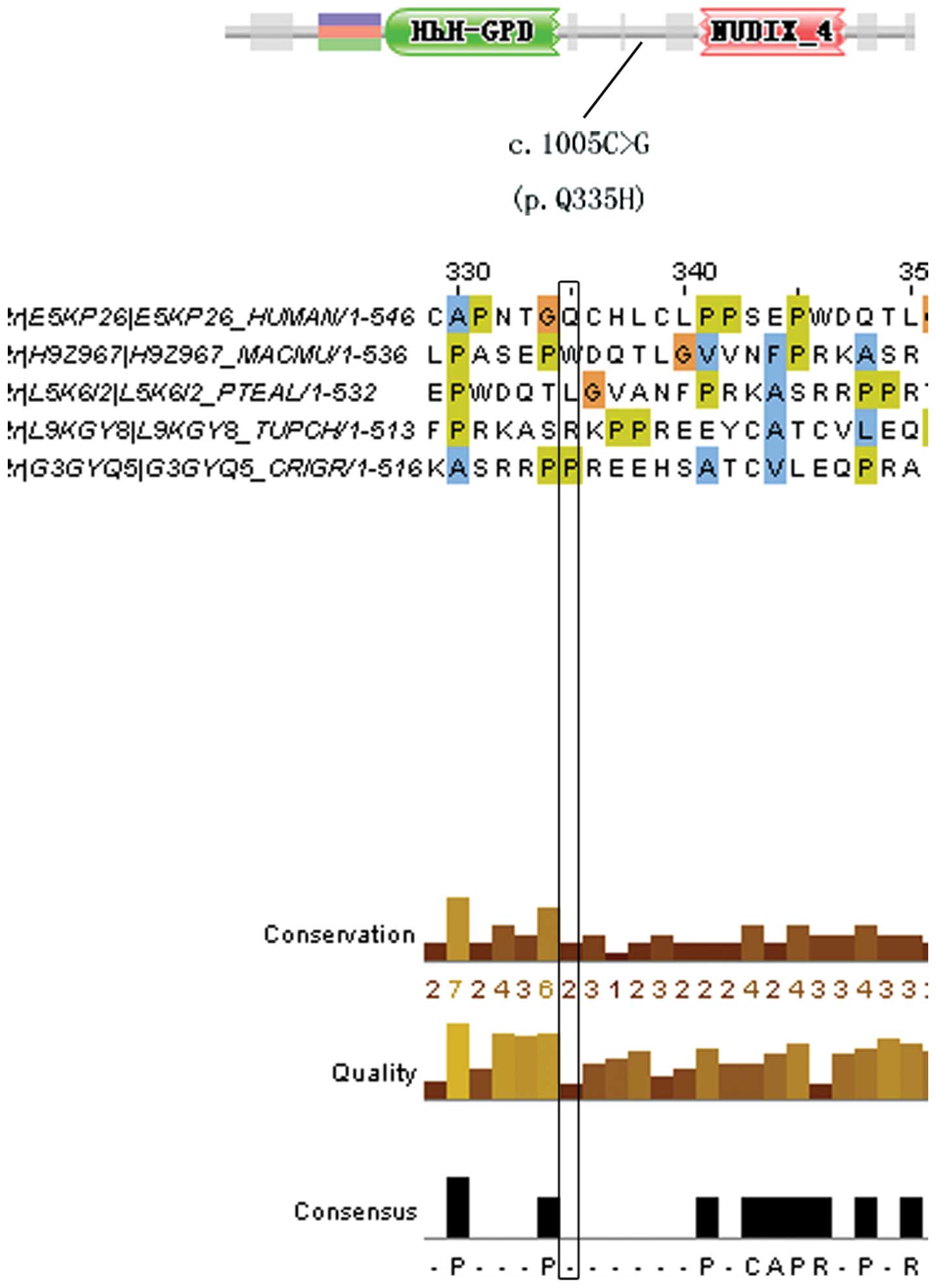
at codon 1196 and generated a premature truncated protein (38). In addition, one missense mutation
(Q335H) and two intronic substitutions (c.264+11G>A and
c.420+35A>G) were identified in the MUTYH gene, and four
synonymous mutations (I144I, P455P, P462P and L688L) and three
intonic mutations (c.1060–77G>T, c.1060–287A>G and
c.1060–282A>G) were identified in the AXIN2 gene.
| Table II.Variants identified in each
patient. |
Table II.
Variants identified in each
patient.
| Gene | Exon or intron | Variant | Function class | Case no. |
|---|
| APC | 11 | c.1458T>C (P.
Y486Y) | Synonymous
substitution | II, III and IV |
|
| 11 | c.1347G>C (p.
T449T) | Synonymous
substitution | III, IV, IX and
XI |
|
| 15 | c.3518T>G (p.
1173V>G) | Missense
substitution | II, IV, V, VIII, IX
and XI |
|
| 15 | c. 3587C>A (p.
S1196X) | Nonsense
substitution | IV |
|
| 15 | c.4425G>A (p.
T1493T) | Synonymous
substitution | III, VIII and
IX |
|
| 15 | c.5034 G>A (p.
G1678G) | Synonymous
substitution | III and IX |
|
| 15 | c.5268T>G (p.
S1756S) | Synonymous
substitution | II, III, IV and
VIII |
|
| 15 | c.5279C>A (p.
1760P>H) | Missense
substitution | II, III and IV |
|
| 15 | c. 5880G>A (p.
P1960P) | Synonymous
substitution | IV, VIII, IX and
XI |
|
| 15 | c.5465T>A (p.
V1822D) | Missense
substitution | I, IV, VI, VII and
X |
|
| 15 | c.6170A>G (p.
K2057R) | Missense
substitution | II, IX and XI |
| MUTYH | 11 | c.1005C>G (p.
Q335H) | Missense
substitution | II, III, V, VIII,
IX and XI |
|
| IVS5 | c.264+11G>A | Intronic
mutation | II, III, V, VIII,
IX and XI |
|
| IVS5 | c.420+35A>G | Intronic
mutation | II, III, V, VIII,
IX and XI |
|
|
|
|
| II, III, V, VIII,
IX and XI |
| AXIN2 | 2 | c.432T>C (p.
I144I) | Synonymous
substitution | II, III, V, VIII,
IX and XI |
|
| 6 | c.1365A>G (p.
P455P) | Synonymous
substitution | II, III, V, VIII,
IX and XI |
|
| 6 | c.1386C>T9 (p.
P462P) | Synonymous
substitution | II, III, V, VIII,
IX and XI |
|
| 8 | c.2062C>T (p.
L688L) | Synonymous
substitution | II, III, V, VIII,
IX and XI |
|
| IVS3 |
c.1060–287A>G | Intronic
mutation | II, III, V, VIII,
IX and XI |
|
| IVS3 |
c.1060–282A>G | Intronic
mutation | II, III, V, VIII,
IX and XI |
|
| IVS3 |
c.1060–77G>T | Intronic
mutation | II, III, V, VIII,
IX and XI |
Computational prediction of missense
substitutions
Among the APC missense substitutions identified in
our study, the functional assessment of V1822D indicated that it is
a deleterious mutation, and the bioinformatic result was consistent
with that of previous studies, which considered it as a high-risk
polymorphism associated with CRC (39–41).
Computational prediction suggested that the missense substitutions
in the APC and MUTYH gene might be disease-associated variants
(Table III, Figs. 1 and 2).
It is of note that the novel APC missense substitution (V1173G),
which mapped to the fourth 15aa repeat region of the APC protein,
an essential region involved in binding with β-catenin, might be
associated with the early onset of FAP (42).
| Table III.Computational predictions of
deleterious missense mutations. |
Table III.
Computational predictions of
deleterious missense mutations.
| Gene | Exon | Variant | dbSNP ID | Function class |
PolyPhen-2a | SIFTb |
SNPs&GOc |
|---|
| APC | 15 | c.3518T>G (p.
1173V>G) | Novel | Missense
mutation | Benign (0.003) | Tolerated
(0.540) | Disease |
| APC | 15 | c.5279C>A (p.
1760P>H) | Novel | Missense
mutation | Benign (0.039) | Tolerated
(0.100) | Disease |
| APC | 15 | c.5465T>A (p.
V1822D) | rs459552 | Missense
mutation | Benign (0.000) | Affected protein
function (0.000) | Disease |
| APC | 15 | c.6170A>G (p.
K2057R) | Novel | Missense
mutation | Benign (0.411) | Tolerated
(0.470) | Disease |
| MUTYH | 11 | c.1005 C>G (p.
Q335H) | rs3219489 | Missense
mutation | Benign (0.124) | Tolerated
(0.140) | Disease |
Splicing regulation of synonymous and
intronic substitutions
Synonymous and intronic substitutions of each gene
were further analyzed (Table IV).
In silico analysis results indicated that the six already
known synonymous substitutions (Y486Y, T449T, T1493T, G1678G,
S1756S and P1960P) might affect splicing regulation by creating or
removing exonic splicing enhancers or exonic splicing silencers;
however, in previous studies of these synonymous substitutions,
they were considered to be ‘silent’ substitutions (43,44). No
effect was detected in the synonymous or intonic substitutions of
the AXIN2 gene.
| Table IV.Computational predictions of the
effect of splicing of variants in each gene. |
Table IV.
Computational predictions of the
effect of splicing of variants in each gene.
| Gene | Exon/intron | Variant | dbSNP ID | Function class | ESEfinder | ESRSearch | PESX | RESCUE_ESE | Splicing
affection |
|---|
| APC | 11 | c.1458T>C (P.
Y486Y) | rs2229992 | Synonymous
mutation | Changed | Changed | Changed | Not changed | Exon skipping |
| APC | 11 | c.1347G>C (p.
T449T) | rs74318065 | Synonymous
mutation | Changed | Changed | Changed | Changed | Exon skipping |
| APC | 15 | c.4425G>A (p.
T1493T) | rs41115 | Synonymous
mutation | Changed | Changed | Changed | Not changed | Exon skipping |
| APC | 15 | c.5034G>A (p.
G1678G) | rs42427 | Synonymous
mutation | Changed | Changed | Changed | Changed | Exon skipping |
| APC | 15 | c.5268T>G (p.
S1756S) | rs866006 | Synonymous
mutation | Changed | Changed | Changed | Changed | Exon skipping |
| APC | 15 | c. 5880G>A (p.
P1960P) | rs465899 | Synonymous
mutation | Changed | Changed | Changed | Changed | Exon skipping |
| MUTYH | IVS 5 | c.264+11G>A | rs139977567 | Intronic
mutation | N/A | N/A | N/A | N/A | No effect |
| MUTYH | IVS 5 | c.420+35A>G | rs3219487 | Intronic
mutation | N/A | N/A | N/A | N/A | No effect |
| AXIN2 | 2 | c.432T>C (p.
I144I) | rs2240307 | Synonymous
mutation | N/A | N/A | N/A | N/A | No effect |
| AXIN2 | 6 | c.1365A>G (p.
P455P) | rs9915936 | Synonymous
mutation | N/A | N/A | N/A | N/A | No effect |
| AXIN2 | 6 | c.1386C>T9 (p.
P462P) | rs1133683 | Synonymous
mutation | N/A | N/A | N/A | N/A | No effect |
| AXIN2 | 8 | c.2062C>T (p.
L688L) | rs35415678 | Synonymous
mutation | N/A | N/A | N/A | N/A | No effect |
| AXIN2 | IVS 3 | c.1060–287
A>G | rs4467119 | Intronic
mutation | N/A | N/A | N/A | N/A | No effect |
| AXIN2 | IVS 3 | c.1060–282
A>G | rs4464120 | Intronic
mutation | N/A | N/A | N/A | N/A | No effect |
| AXIN2 | IVS 3 |
c.1060–77G>T | rs4541111 | Intronic
mutation | N/A | N/A | N/A | N/A | No effect |
Discussion
Due to the poor prognosis of FAP and its genetic
diversity, clinicians and researchers study FAP from various
angles, each having their own agenda. Recent advances in our
understanding of FAP suggest that the genetics of each patient
might help to clarify diagnosis, lead to early cancer surveillance
options, and guide surgical and chemopreventive management for FAP
patients. However, the genetics of FAP vary markedly among
different countries (9–15). FAP in China has its own
characteristics (6–9,45), which
may be explained by the abundant genetic resources in 56
nationalities as well as religious differences. The present study
is a comprehensive mutational analysis of FAP in Yunnan province in
southwest China. Indigenous people in this area live in a
relatively closed environment; thus, 11 unrelated FAP patients from
this area were analyzed to screen for pathogenic changes.
It is well known that FAP is linked to germline
mutations in the APC gene (4,5). In our study, mutational analysis of the
APC gene was firstly conducted, and one known missense mutation
(V1822D) and one previously identified nonsense mutation (S1196X)
were observed. The missense mutation (V1822D) was identified in
five FAP patients in our research indices, and it was established
that the missense substitution is a high-risk polymorphism
associated with CRC (39–41). In addition, a nonsense mutation
(S1196X) was identified in one of the five patients with the
missense mutation V1822D. The nonsense mutation S1196X was reported
to be a pathogenic change predisposing to FAP (38), which results in a stop codon at codon
1196 and generates a premature truncated protein. Additionally,
certain synonymous mutations of the APC gene were identified in the
different FAP patients; six different synonymous mutations (Y486Y,
T449T, T1493T, G1678G, S1756S and P1960P) were identified in all
samples, which were considered as silent mutations in the past. By
referring to previous studies and databases (22,38–41), we
were able to identify pathogenic mutations in 5 of the 11 index
cases in our series, and the detection rate was 54.5%.
The detection rate of APC mutations in FAP patients
varies significantly, ranging from 39 to 90% (10–15). The
mutation negativity of the APC gene may suggest either that APC has
alterations that escape detection by routine techniques, or
alternatively, that other (known or unknown) genes are involved in
FAP predisposition (18). Among the
known genes, the identification of germline mutations in MUTYH and
AXIN2 involved in FAP predisposition have been reported (16–19).
Therefore, in the remaining six patients without already known
pathogenic changes involved in FAP (patients presenting APC
synonymous mutations), whole-gene sequencing of the MUTYH gene and
AXIN2 gene was further performed. The results indicated that two
intronic substitutions (c.264+11G>A and c.420+35A>G) and one
missense mutation (Q335H) were present in the MUTYH gene, and four
synonymous mutations (I144I, P455P, P462P and L688 L) and three
intonic mutations (c.1060–77G>T, c.1060–287A>G and c.1060–282
A>G) were present in the AXIN2 gene.
Like the synonymous mutations observed in APC, these
findings in MUTYH and AXIN2 are still of little use for the genetic
diagnosis of FAP. However, the repeated synonymous and intronic
mutations in the different patients are of note. Traditionally,
synonymous substitutions and certain intronic mutations were
assumed not to cause significant changes in the function of coded
proteins. However, this idea changed with the observation that
silent mutations may cause human diseases (46,47), and
numerous cancer-associated synonymous substitutions have been
reported (48). Accumulated evidence
suggests that certain synonymous and intronic mutations may result
in altered splicing, which may directly cause disease, modify the
severity of the disease phenotype, or be linked with disease
susceptibility (49–52). Approximately 15% of disease-causing
point mutations affect pre-mRNA splicing (53), resulting in the activation of cryptic
sites, creation of a pseudo-exon within an intron, intron
retention, or exon skipping, the latter being the most likely of
these events.
With the rapid development of science and
technology, numerous tools and databases for the prediction of
pathogenicity have been promoted. Using function assessment tools
and databases, we further assessed the polymorphisms in each gene,
and the results indicated that the synonymous substitutions in the
APC gene might be disease-causing point mutations by affecting
pre-mRNA splicing and resulting in the pathogenicity of FAP, while
the mutations identified in the MUTYH gene and AXIN2 gene
demonstrated no impact on predisposing to FAP. Computational
prediction suggested that the missense substitutions in the APC
gene and MUTYH gene might also be disease-associated variants. It
is also worth noting that one of the novel missense substitutions
(V1173G) was identified in APC, which was also predicted to be
associated with the early onset of FAP.
In our study, following mutation analysis of each
gene with conventional methods, we were able to determine the
genetic pathogenicity of five FAP patients, with the remaining six
patients having no known pathogenic changes involved in FAP. Using
function assessment tools and databases, the synonymous mutations
of the APC gene were predicted to affect splicing by creating or
removing exonic splicing enhancers or exonic splicing silencers,
which might result in a pseudo-exon within an intron, intron
retention, or exon skipping. Computational prediction of synonymous
substitutions suggested that certain synonymous mutations of the
APC gene may have a functional impact on the splicing regulation of
APC at the mRNA level. Moreover, the missense substitutions in APC
and MUTYH were also predicted to be disease-associated variants.
These cancer-associated substitutions might explain the
pathogenicity of the other six FAP patients in our study, who were
previously presenting undetectable pathogenic changes.
In conclusion, based on our study, we consider that
it is more beneficial to explore the potential function of the
various polymorphisms than to identify rare variants predisposing
to FAP. The functional prediction of the mutations is a
cost-effective way to identify candidate disease variants in
developing countries (54). Further
research is required from ex vivo splicing assay and
ESE-dependent splicing assay with regard to the synonymous
mutations (51). Due to the high cost
and the time-consuming process of screening the genetic
pathogenicity of FAP, the detection of point mutations by DNA
analysis followed by prediction of the pathogenicity may be a
useful new strategy to provide proper diagnosis or genetic
counseling to patients and their families. This is likely to
contribute to better genetic counseling and simplify the mutation
screening strategy.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81160245) and the Science and
Technology Planning Project of Yunnan Province, China (no.
2011FB160).
References
|
1
|
Gardner EJ, Burt RW and Freston JW:
Gastrointestinal polyposis: syndromes and genetic mechanisms. West
J Med. 132:488–499. 1980.PubMed/NCBI
|
|
2
|
Bianchi LK, Buerke CA, Bennett AE, Lopez
R, Hasson H and Church JM: Fundic gland polyp dysplasia is common
in familial adenomatous polyposis. Clin Gastroenterol Heptatol.
6:180–185. 2008. View Article : Google Scholar
|
|
3
|
Nugent KP and Philips RK: Rectal cancer
risk in older patients with adenomatous polyposis and ileorectal
anastomosis: a cause for concern. Br J Surg. 79:1204–1206. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kinzler KW, Nilbert MC, Vogelstein B,
Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hamilton SR, Hedge P
and Markham A: Identification of a gene located at chromosome 5q21
that is mutated in colorectal cancers. Science. 251:1366–1370.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Groden J, Thliveris A, Samowitz W, Carlson
M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L and
Robertson M: Identification and characterization of the familial
adenomatous polyposis coli gene. Cell. 66:589–600. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu XR, Shan XN, Friedl W, Uhlhaas S, Li
JT, Propping P and Wang YP: Detection of germline mutations in the
APC gene with the protein truncation test. Yi Chuan Xue Bao.
32:903–908. 2005.(In Chinese). PubMed/NCBI
|
|
7
|
Gan Y, Zheng S and Cai X: Detection of a
gene mutation in familial adenomatous polyposis families by
PCR-RFLP method. Zhonghua Yi Xue Za Zhi. 74:352–354. 1994.(In
Chinese). PubMed/NCBI
|
|
8
|
Chen S, Zhou J, Zhang X, Zhou X, Zhu M,
Zhang Y, Ma G and Li J: Mutation analysis of the APC gene in a
Chinese FAP pedigree with unusual phenotype. ISRN Gastroenterol.
2011:9091212011.PubMed/NCBI
|
|
9
|
Sheng JQ, Cui WJ, Fu L, Jin P, Han Y, Li
SJ, Fan RY, Li AQ, Zhang MZ and Li SR: APC gene mutations in
Chinese familial adenomatous polyposis patients. World J
Gastroenterol. 16:1522–1526. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vandrovcová J, Štekrová J, Kebrdlová V and
Kohoutová M: Molecular analysis of the APC and MYH genes in Czech
families affected by FAP or multiple adenomas: 13 novel mutations.
Hum Mutat. 23:3972004. View Article : Google Scholar
|
|
11
|
Gómez-Fernández N, Castellví-Bel S,
Fernández-Rozadilla C, Balaguer F, Muñoz J, Madrigal I, Milà M,
Graña B, Vega A, Castells A, et al: Molecular analysis of the APC
and MUTYH genes in Galician and Catalonian FAP families: a
different spectrum of mutations? BMC Med Genet. 10:572009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rivera B, González S, Sánchez-Tomé E,
Blanco I, Mercadillo F, Letón R, Benítez J, Robledo M, Capellá G
and Urioste M: Clinical and genetic characterization of classical
forms of familial adenomatous polyposis: a Spanish population
study. Ann Oncol. 22:903–909. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fostira F, Thodi G, Sandaltzopoulos R,
Fountzilas G and Yannoukakos D: Mutational spectrum of APC and
genotype-phenotype correlations in Greek FAP patients. BMC Cancer.
10:3892010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Torrezan GT, da Silva FC, Santos EM,
Krepischi AC, Achatz MI, Aguiar S Jr, Rossi BM and Carraro DM:
Mutational spectrum of the APC and MUTYH genes and
genotype-phenotype correlations in Brazilian FAP, AFAP and MAP
patients. Orphanet J Rare Dis. 8:542013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanter-Smoler G, Fritzell K, Rohlin A,
Engwall Y, Hallberg B, Bergman A, Meuller J, Grönberg H, Karlsson
P, Björk J and Nordling M: Clinical characterization and the
mutation spectrum in Swedish adenomatous polyposis families. BMC
Med. 6:102008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Al-Tassan N, Chmiel NH, Maynard J, Fleming
N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS,
Sampson JR and Cheadle JP: Inherited variants of MYH associated
with somatic G:C->T:A mutations in colorectal tumors. Nat Genet.
30:227–232. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sieber OM, Lipton L, Crabtree M, Heinimann
K, Fidalgo P, Phillips RK, Bisgaard ML, Orntoft TF, Aaltonen LA,
Hodgson SV, et al: Multiple colorectal adenomas, classic
adenomatous polyposis and germ-line mutations in MYH. N Engl J Med.
348:791–799. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Renkonen ET, Nieminen P, Abdel-Rahman WM,
Moisio AL, Järvelä I, Arte S, Järvinen HJ and Peltomäki P:
Adenomatous polyposis families that screen APC mutation-negative by
conventional methods are genetically heterogeneous. J Clin Oncol.
23:5651–5659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lammi L, Arte S, Somer M, Jarvinen H,
Lahermo P, Thesleff I, Pirinen S and Nieminen P: Mutations in AXIN2
cause familial tooth agenesis and predispose to colorectal cancer.
Am J Hum Genet. 74:1043–1050. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei SC, Su YN, Tsai-Wu JJ, Wu CH, Huang
YL, Sheu JC, Wang CY and Wong JM: Genetic analysis of the APC gene
in Taiwanese familial adenomatous polyposis. J Biomed Sci.
11:260–265. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Prior TE and Bridgeman SJ: Identifying
mutations for MYH-associated polyposis. Current Protocols in Human
Genetics 64: 10.13.1–10.13.14. January 1–2010.DOI:
10.1002/0471142905.hg1013s64. View Article : Google Scholar
|
|
22
|
Pan M, Cong P, Wang Y, Lin C, Yuan Y, Dong
J, Banerjee S, Zhang T, Chen Y, Zhang T, et al: Novel LOVD
databases for hereditary breast cancer and colorectal cancer genes
in the Chinese population. Hum Mutat. 32:1335–1340. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang T, Moss A, Cong P, Pan M, Chang B,
Zheng L, Fang Q, Zareba W, Robinson J, Lin C, et al: Long QT
International Registry Investigators; HVP-China Investigators: LQTS
gene LOVD database. Hum Mutat. 31:E1801–E1810. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: DbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hubbard TJ, Aken BL, Beal K, Ballester B,
Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, et
al: Ensembl 2007. Nucleic Acids Res. 35(Database issue): D610–D617.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Calabrese R, Capriotti E, Fariselli P,
Martelli PL and Casadio R: Functional annotations improve the
predictive score of human disease-related mutations in proteins.
Hum Mutat. 30:1237–1244. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Capriotti E and Altman RB: Improving the
prediction of disease-related variants using protein
three-dimensional structure. BMC Bioinformatics. 12(Suppl 4):
S32011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Capriotti E, Calabrese R, Fariselli P,
Martelli PL, Altman RB and Casadio R: WS-SNPs&GO: a web server
for predicting the deleterious effect of human protein variants
using functional annotation. BMC Genomics. 14(Suppl 3): S62013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cartegni L, Wang J, Zhu Z, Zhang MQ and
Krainer AR: ESEfinder: a web resource to identify exonic splicing
enhancers. Nucleic Acids Res. 31:3568–3571. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Punta M, Coggill PC, Eberhardt RY, Mistry
J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et
al: The Pfam protein families database. Nucleic Acids Res.
40(Database issue): D290–D301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Larkin MA, Blackshields G, Brown NP,
Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm
A, Lopez R, et al: Clustal W and Clustal X version 2.0.
Bioinformatics. 23:2947–2948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Waterhouse AM, Procter JB, Martin DM,
Clamp M and Barton GJ: Jalview Version 2 - a multiple sequence
alignment editor and analysis workbench. Bioinformatics.
25:1189–1191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yeo G, Burge CB, Hoon S and Venkatesh B:
Variation in sequence and organization of splicing regulatory
elements in vertebrate genes. Proc Natl Acad Sci USA.
101:15700–15705. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fairbrother WG, Yeh RF, Sharp PA and Burge
CB: Predictive identification of exonic splicing enhancers in human
genes. Science. 297:1007–1013. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang XH, Kangsamaksin T, Chao MS,
Banerjee JK and Chasin LA: Exon inclusion is dependent on
predictable exonic splicing enhancers. Mol Cell Biol. 25:7323–7332.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Friedl W and Aretz S: Familial adenomatous
polyposis: experience from a study of 1164 unrelated german
polyposis patients. Hered Cancer Clin Pract. 3:95–114. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liang J, Lin C, Hu F, Wang F, Zhu L, Yao
X, Wang Y and Zhao Y: APC polymorphisms and the risk of colorectal
neoplasia: a HuGE review and meta-analysis. Am J Epidemiol.
177:1169–1179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guerreiro CS, Cravo ML, Brito M, Vidal PM,
Fidalgo PO and Leitão CN: The V1822D APC polymorphism interacts
with fat, calcium and fiber intakes in modulating the risk of
colorectal cancer in Portuguese persons. Am J Clin Nutr.
85:1592–1597. 2007.PubMed/NCBI
|
|
41
|
Menéndez M, González S, Blanco I, Guinó E,
Peris M, Peinado MA, Capellá G and Moreno V: Colorectal cancer risk
and the APC V1822D variant. Int J Cancer. 112:161–163. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nakagawa H, Murata Y, Koyama K, Fujiyama
A, Miyoshi Y, Monden M, Akiyama T and Nakamura Y: Identification of
a brain-specific APC homologue, APCL and its interaction with
beta-catenin. Cancer Res. 58:5176–5181. 1998.PubMed/NCBI
|
|
43
|
Wachsmannova-Matelova L, Stevurkova V,
Adamcikova Z, Holec V and Zajac V: Polymorphisms in the adenomatous
polyposis coli gene in Slovak families suspected of FAP. Neuro
Endocrinol Lett. 30(Suppl 1): S25–S28. 2009.
|
|
44
|
Herrmann SM, Adler YD, Schmidt-Petersen K,
Nicaud V, Morrison C, Paul M and Zouboulis ChC: The concomitant
occurrence of multiple epidermal cysts, osteomas and thyroid gland
nodules is not diagnostic for Gardner syndrome in the absence of
intestinal polyposis: a clinical and genetic report. Br J Dermatol.
149:877–883. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shu Z, Yanqin H and Ying Y: Hereditary
colorectal cancer in china. Hered Cancer Clin Pract. 3:155–164.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cartegni L, Chew SL and Krainer AR:
Listening to silence and understanding nonsense: exonic mutations
that affect splicing. Nat Rev Genet. 3:285–298. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pagani F and Baralle FE: Genomic variants
in exons and introns: Identifying the splicing spoilers. Nat Rev
Genet. 5:389–396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Grosso AR, Martins S and Carmo-Fonseca M:
The emerging role of splicing factors in cancer. EMBO Rep.
9:1087–1093. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bechtel JM, Rajesh P, Ilikchyan I, Deng Y,
Mishra PK, Wang Q, Wu X, Afonin KA, Grose WE, Wang Y, et al: The
Alternative Splicing Mutation Database: a hub for investigations of
alternative splicing using mutational evidence. BMC Res Notes.
1:32008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Covaciu C, Grosso F, Pisaneschi E,
Zambruno G, Gregersen PA, Sommerlund M, Hertz JM and Castiglia D: A
founder synonymous COL7A1 mutation in three Danish families with
dominant dystrophic epidermolysis bullosa pruriginosa identifies
exonic regulatory sequences required for exon 87 splicing. Br J
Dermatol. 165:678–682. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tournier I, Vezain M, Martins A,
Charbonnier F, Baert-Desurmont S, Olschwang S, Wang Q, Buisine MP,
Soret J, Tazi J, et al: A large fraction of unclassified variants
of the mismatch repair genes MLH1 and MSH2 is associated with
splicing defects. Hum Mutat. 29:1412–1424. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pagani F, Raponi M and Baralle FE:
Synonymous mutations in CFTR exon 12 affect splicing and are not
neutral in evolution. Proc Natl Acad Sci USA. 102:6368–6372. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Krawczak M, Reiss J and Cooper DN: The
mutational spectrum of single base-pair substitutions in mRNA
splice junctions of human genes: causes and consequences. Hum
Genet. 90:41–54. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nouri N, Fazel-Najafabadi E, Behnam M,
Nouri N, Aryani O, Ghasemi M, Nasiri J and Sedghi M: Use of in
silico tools for classification of novel missense mutations
identified in dystrophin gene in developing countries. Gene.
535:250–254. 2014. View Article : Google Scholar : PubMed/NCBI
|