Introduction
Among women, cervical cancer is the second most
commonly diagnosed type of cancer and the third leading cause of
cancer-associated mortality (1).
Worldwide, ~500,000 cervical cancer cases are diagnosed and 230,000
mortalities occur due to this disease annually (2). DNA methylation, a type of epigenetic
event, may increase the risk of cervical cancer through regulating
gene expression and chromatin structure (3). Methylated carcinogenic human
papillomavirus (HPV) DNA may be used as a predictive and/or
diagnostic biomarker for risk of cervical cancer (4). Sood et al (3) demonstrated that methylation of the genes
myogenic differentiation 1, telomerase reverse transcriptase and
Ras association domain family member 1 could predict a more
favorable outcome in patients with invasive cervical carcinoma
treated with standard chemoradiation therapy. Kalantari et
al (5) reported that methylation
of L2 and L1 genes in HPV16, 18, 31 and 45, and of the cellular
death-associated protein kinase gene were considered biomarkers of
the progression of cervical neoplasia. Furthermore, Nye et
al (6) revealed that infection
with high-risk HPV types was associated with differentially
methylated regions in the paternally expressed 3 (PEG3)
gene, and that PEG3 methylation status may have potential as
a molecular marker for screening of cervical intraepithelial
neoplasia. Numerous studies have reported that methylation of
certain genes is associated with the pathogenesis of cervical
cancer, while the mechanisms of development and progression of
cervical cancer remain unclear (7,8).
Recently, microarray analyses have been performed to
identify gene methylation, gene expression and RNA regulation in
cervical cancer. Sun et al (9)
analyzed differentially expressed long non-coding RNAs (lncRNAs)
and mRNAs in the microarray data set GSE55940, and revealed that
the enhancer of zeste 2 polycomb repressive complex 2 subunit
(EZH2) gene and EZH2-binding lncRNA in cervical cancer
(lncRNA-EBIC) may play roles in the repression of E-cadherin, which
could contribute to the metastasis of cervical cancer. Ye et
al (10) also analyzed the data
in GSE55940, and determined that microRNA-145 expression was
decreased in cervical cancer tissues, and was associated with
advanced cancer stages and moderate/poor differentiation. In
addition, Burris et al (11)
analyzed GSE46306 and revealed that cervical DNA methylation of the
prostaglandin E receptor 2 and long interspersed nuclear element-1
Homo sapiens-specific genes was associated with the length
of gestation in humans. Although many genes are associated with the
progression of cervical cancer, comprehensive analyses of
transcriptome microarrays and methylation microarrays have rarely
been reported.
It is of interest to further explore the mechanisms
of cervical cancer in a comprehensive manner. In the current study,
a comprehensive analysis of the GSE55940 and GSE46306 data sets was
performed to identify genes involved in the pathogenesis of
cervical cancer. Overlapping differentially expressed genes (DEGs)
in GSE55940 and differentially methylated genes in GSE46306 were
identified, and Gene Ontology (GO) enrichment analysis for these
genes was performed.
Materials and methods
Data preprocessing
GSE55940 was downloaded from the Gene Expression
Omnibus database (GEO; http://www.ncbi.nlm.nih.gov/geo/); this data set
included 5 separate cervical cancer tissues and 5 paired adjacent
non-tumor samples. The platform of GSE55940 was GPL16238:
[hGlue_3_0_v1] Glue Grant Human Transcriptome Array version 3.0
(GG-H) [transcript-level]. GSE46306 was also downloaded from the
GEO database. In this study, 20 normal cervical samples
(HPV-negative) and 6 cervical cancer tissues (HPV-positive) were
included in the subsequent analysis. GPL13534 Illumina
HumanMethylation450 BeadChip (HumanMethylation450_15017482) was
used to detect the methylation level of the CpG sites in
GSE46306.
GSE55940 was preprocessed using affy package
(www.bioconductor.org/packages/release/bioc/html/affy.html)
in R language. Background correction, quantile normalization and
probe summarization were performed using the Robust Multi-array
Average algorithm. For the data set GSE46306, the CpG sites with
P>0.05 and the samples [CpG sites (P>0.05)/all CpG sites
>1/100,000] were first excluded. Peak standardization was then
performed for the remaining CpG sites and samples using the
Illumina Methylation Analyzer (IMA) package (12).
Identification of DEGs and
differentially methylated genes
The normalized data GSE55940 were further analyzed
by Limma package (13), and genes
with P<0.05 and |log[fold change (FC)]|>0.5 were defined as
DEGs.
For the data GSE46306, the differentially methylated
genes were identified based on three levels: CpG site level, CpG
island level and gene level. For CpG site level, the average β
value (βa) in normal cervical and cervical cancer
samples was calculated for every CpG site. Limma method in IMA was
used to identify differentially methylated CpG sites that met the
criteria |∆βa|>0.2 and P<0.05. Genes containing
the differentially methylated sites were obtained based on the
annotation file of the microarray platform.
For CpG island level, the β values of five region
categories [CpG islands, upstream 2,000 bp of CpG islands
(N-shore), upstream 2,001–4,000 bp of CpG islands (N-shelf),
downstream 2,000 bp of CpG islands (S-shore), and upstream
2,001–4,000 bp of CpG islands (S-shelf)] were calculated based on
the βa value of the CpG sites in those regions, and
opensea referred to the annotations associated with UCSC islands.
Additionally, Limma method in IMA was used to identify
differentially methylated regions with the cut-off of |∆β|>0.2
and P<0.05. The genes located in the differentially methylated
regions were obtained based on the annotation file of the
microarray platform.
For the gene level, the β values of the upstream 200
bp of genes (TSS200), the upstream 201–1,500 bp of genes (TSS1500),
5′ untranslated region (UTR), the first exon region, the gene body
regions and 3′-UTR were calculated using a t-test, and the
gene regions with and |∆β|>0.2 and P<0.05 were considered to
be differentially methylated.
Comprehensive analysis of DEGs and
differentially methylated genes
For GSE46306, the overlapping genes that were
identified based on CpG site level, CpG island level and gene level
were screened. Subsequently, the overlapping genes of the
overlapping differentially methylated genes in GSE46306 and the
DEGs in GSE55940 were also screened, and GO enrichment analysis was
performed for these genes using the ToppGene database (toppgene.cchmc.org/).
Results
Analysis of DEGs
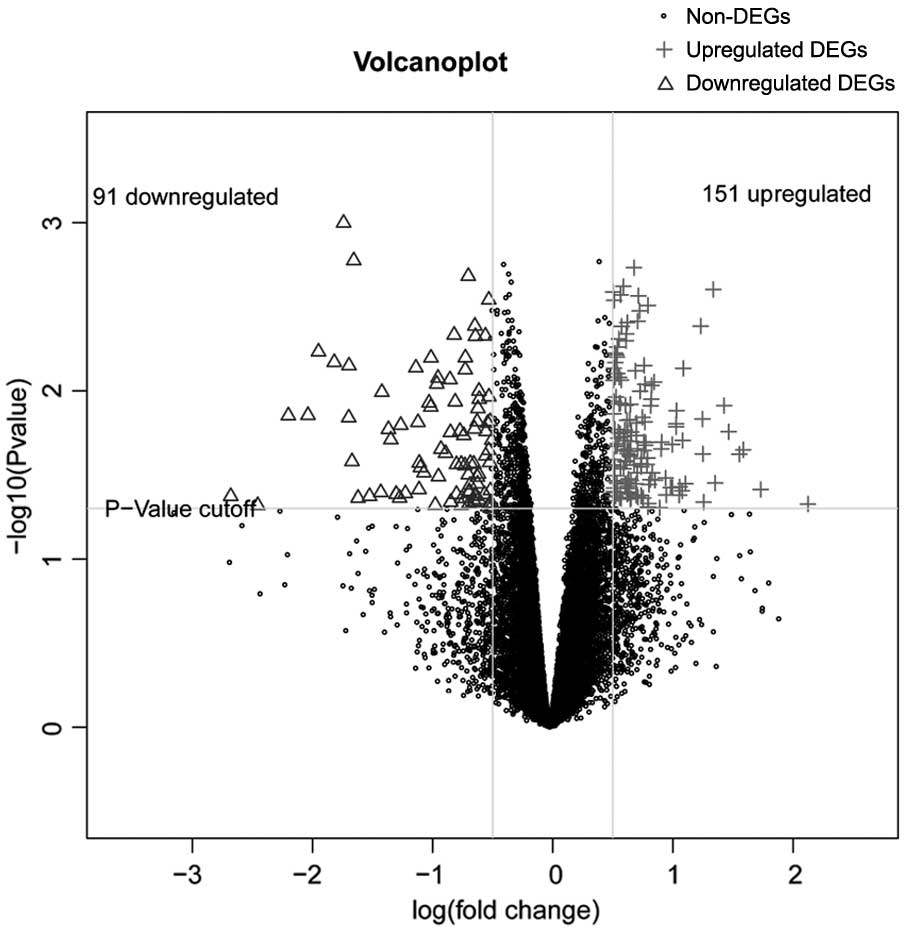
In the analysis of GSE55940, a total of 242 DEGs
between the cervical cancer group and the adjacent non-tumor group
were identified, including 91 downregulated and 151 upregulated
DEGs. The distribution of DEGs is shown in Fig. 1.
Analysis of differentially methylated
genes
For GSE46306, 15,515 differentially methylated CpG
sites were identified, involving 3,064 genes, 2,772 differentially
methylated CpG island regions containing 1,082 genes, and 1,857
differentially methylated gene regions that related to 1,012 genes,
between the cervical cancer group and the control group (Table I). Distribution locations and
percentages of differentially methylated regions at the CpG island
level are shown in Fig. 2; the
distribution was 52.5% CpG islands, 16.9% N-shore, 16.7% opensea,
10.8% S-shore, 1.6% N-shelf and 1.5% S-shelf. In addition, the
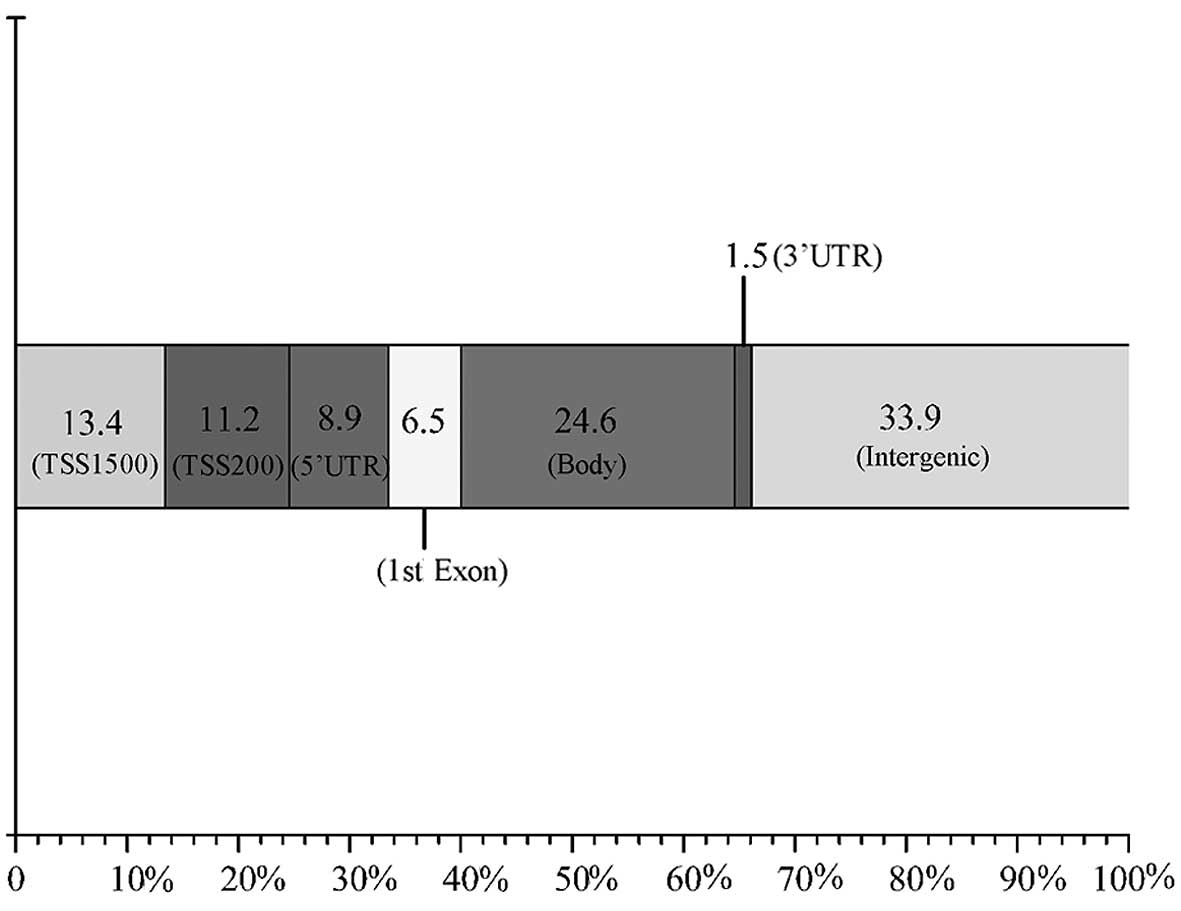
distribution locations and percentages of differentially methylated
regions at the gene level are shown in Fig. 3, comprising 33.9% intergenic, 24.6%
gene body, 13.4% TSS1500, 11.2% TSS200, 8.9% 5′-UTR, 6.5% 1st exon
and 1.5% 3′-UTR.
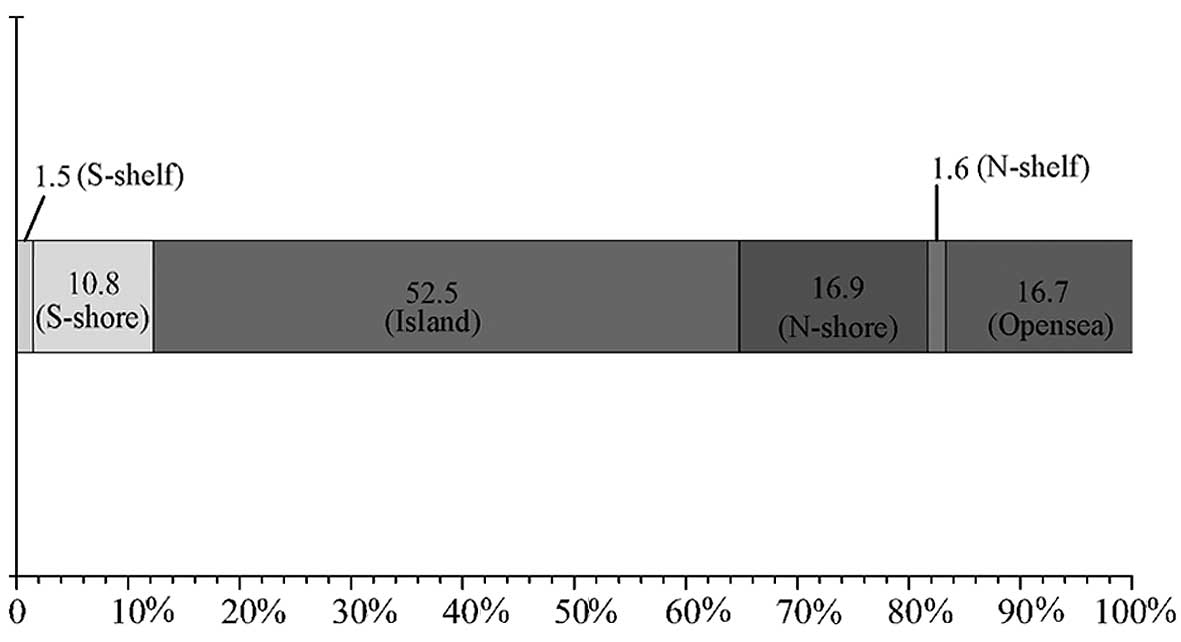 | Figure 2.Distribution locations and
percentages of differentially methylated regions between the
cervical cancer group and the control group at CpG island level.
S-shelf, upstream 2,001–4,000 bp of CpG islands; S-shore,
downstream 2,000 bp of CpG islands; N-shore, upstream 2,000 bp of
CpG islands; N-shelf, upstream 2,001–4,000 bp of CpG islands. |
 | Table I.Number of differentially methylated
CpG sites, differentially methylated regions at the CpG island
level and differentially methylated regions at the gene level
between the cervical cancer group and the control group. |
Table I.
Number of differentially methylated
CpG sites, differentially methylated regions at the CpG island
level and differentially methylated regions at the gene level
between the cervical cancer group and the control group.
| Type | Region | Count | Gene count |
|---|
| CpG sites | Whole genome | 15,515 | 3,064 |
| Gene region | TSS1500 | 259 | 1,012 |
|
| TSS200 | 497 |
|
|
| 5′-UTR | 382 |
|
|
| 3′-UTR | 91 |
|
|
| Gene body | 121 |
|
|
| Exon 1 | 507 |
|
| CpG island
region | Island | 1,505 | 1,082 |
|
| N-shore | 607 |
|
|
| N-shelf | 78 |
|
|
| S-shore | 520 |
|
|
| S-shelf | 62 |
|
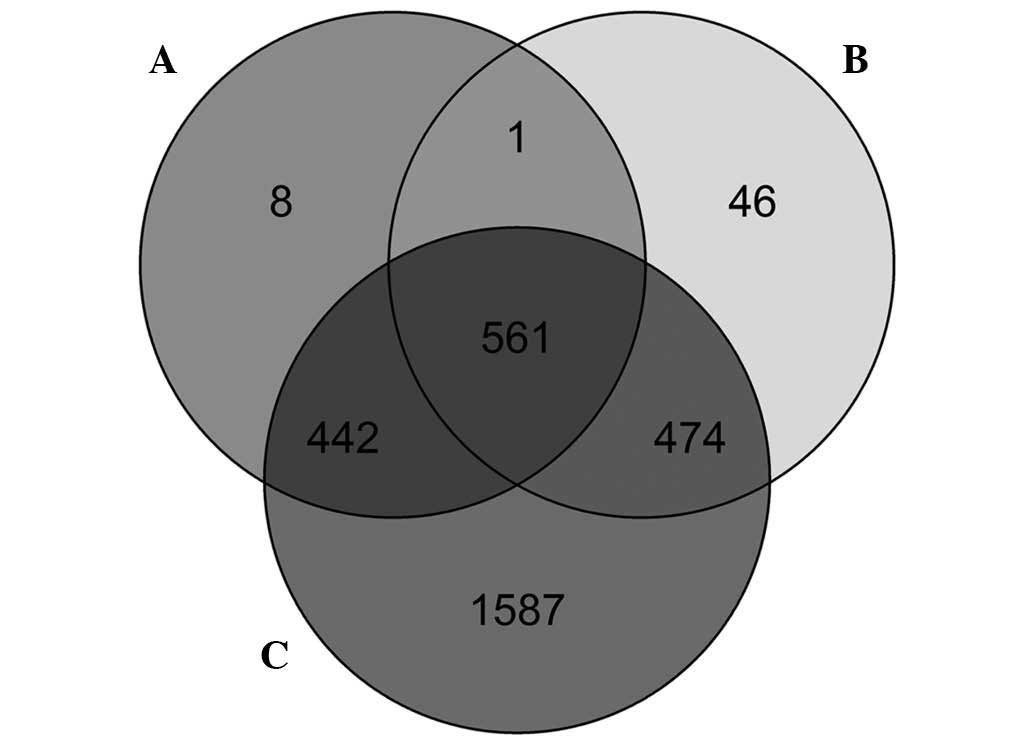
In GSE46306, 561 overlapping differentially
methylated genes were identified through the differential
methylation analysis at the CpG site level, CpG island level and
gene level (Fig. 4). In addition, 5
overlapping genes between the 561 overlapping differentially
methylated genes and the 242 DEGs were identified (Table II): dipeptidyl peptidase 4
(DPP4); endothelin 3 (EDN3); fibroblast growth factor
14 (FGF14); tachykinin, precursor 1 (TAC1); and
wingless-type MMTV integration site family, member 16
(WNT16). All of the 5 genes were downregulated and
hypermethylated in cervical cancer samples.
| Table II.Overlapping genes between the 561
overlapping differentially methylated genes in GSE46306 and 242
differentially expressed genes in GSE55940. |
Table II.
Overlapping genes between the 561
overlapping differentially methylated genes in GSE46306 and 242
differentially expressed genes in GSE55940.
| Gene | logFC | CpG ID | ∆β |
|---|
| DPP4 | −0.567 | cg09601770 | 0.210 |
|
|
| cg10112391 | 0.208 |
|
|
| cg12335708 | 0.212 |
|
|
| cg18892517 | 0.234 |
|
|
| cg19350270 | 0.274 |
|
|
| cg25912827 | 0.248 |
| EDN3 | −1.227 | cg04048259 | 0.345 |
|
|
| cg08318212 | 0.238 |
|
|
| cg09005679 | 0.286 |
|
|
| cg13919285 | 0.213 |
|
|
| cg16205854 | 0.226 |
|
|
| cg17146570 | 0.231 |
|
|
| cg20910807 | 0.233 |
|
|
| cg21163415 | 0.395 |
|
|
| cg21512644 | 0.325 |
| FGF14 | −0.558 | cg02491276 | 0.202 |
|
|
| cg05210258 | 0.377 |
|
|
| cg08597761 | 0.410 |
|
|
| cg09896622 | 0.277 |
|
|
| cg16398329 | 0.259 |
|
|
| cg20335672 | 0.244 |
|
|
| cg22583065 | 0.245 |
|
|
| cg22809871 | 0.292 |
|
|
| cg23809442 | 0.215 |
|
|
| cg24172416 | 0.280 |
|
|
| cg25317585 | 0.370 |
| TAC1 | −0.648 | cg01287975 | 0.359 |
|
|
| cg09236284 | 0.253 |
|
|
| cg10997627 | 0.244 |
|
|
| cg11873482 | 0.333 |
|
|
| cg16288089 | 0.297 |
|
|
| cg17437939 | 0.387 |
|
|
| cg19212224 | 0.276 |
| WNT16 | −0.728 | cg05470554 | 0.273 |
|
|
| cg14448169 | 0.265 |
|
|
| cg16868298 | 0.274 |
|
|
| cg18579879 | 0.223 |
|
|
| cg25608490 | 0.235 |
|
|
| cg26690075 | 0.280 |
Enrichment analysis of DEGs
The genes DPP4, EDN3, FGF14, TAC1 and
WNT16 were enriched in 86 GO terms. The top ten GO
enrichment terms are shown in Table
III. In particular, these genes were predominantly enriched in
cell migration and cell proliferation, including receptor binding
(DPP4, EDN3, FGF14, TAC1 and WNT16), ameboidal-type
cell migration (DPP4, EDN3 and TAC1),
mitogen-activated protein kinase (MAPK) cascade (FGF14, EDN3
and WNT16) and cell proliferation (EDN3, WNT16, DPP4
and TAC1).
| Table III.Top ten GO terms of DPP4,
EDN3, FGF14, TAC1 and WNT16. |
Table III.
Top ten GO terms of DPP4,
EDN3, FGF14, TAC1 and WNT16.
| Category | ID | Terms | P-value | Count | Genes |
|---|
| GO: MF | GO:0005102 | Receptor
binding |
2.63×10−6 | 5 | FGF14,
EDN3, WNT16, DPP4, TAC1 |
| GO: BP | GO:0001667 | Ameboidal-type cell
migration |
2.66×10−5 | 3 | EDN3,
DPP4, TAC1 |
| GO: BP | GO:0002027 | Regulation of heart
rate |
1.79×10−4 | 2 | EDN3,
TAC1 |
| GO: BP | GO:0050886 | Endocrine
process |
2.22×10−4 | 2 | EDN3,
TAC1 |
| GO: BP | GO:0046887 | Positive regulation
of hormone secretion |
3.73×10−4 | 2 | EDN3,
TAC1 |
| GO: BP | GO:0000165 | Mitogen-activated
protein kinase cascade |
4.16×10−4 | 3 | FGF14,
EDN3, WNT16 |
| GO: BP | GO:0007610 | Behavior |
4.84×10−4 | 3 | FGF14,
EDN3, TAC1 |
| GO: BP | GO:0023014 | Signal transduction
by phosphorylation |
4.96×10−4 | 3 | FGF14,
EDN3, WNT16 |
| GO: BP | GO:0008283 | Cell
proliferation |
4.98×10−4 | 4 | EDN3,
WNT16, DPP4, TAC1 |
| GO: BP | GO:0051050 | Positive regulation
of transport |
5.42×10−4 | 3 | FGF14,
EDN3, TAC1 |
Discussion
DNA methylation is one of the most common epigenetic
events and can regulate the expression of certain genes through
preventing the binding of transcription factors with the genes
(14). A number of studies have
investigated the relationship between the occurrence and
development of cervical cancer and DNA methylation or gene
expression (15,16). However, comprehensive studies of DNA
methylation and gene expression are rare.
In the current study, 5 genes (DPP4,
EDN3, FGF14, TAC1, and WNT16) that were
overlapping between the 561 overlapping differentially methylated
genes and the 242 DEGs were identified; these genes were
downregulated and hypermethylated simultaneously in cervical cancer
samples. The predominant enriched GO terms included receptor
binding, ameboidal-type cell migration, MAPK cascade and cell
proliferation. The comprehensive analysis of transcriptome and
methylation microarrays indicated that DPP4, EDN3,
FGF14, TAC1 and WNT16 may contribute to the
development of cervical cancer.
DPP4 [also known as cluster of
differentiation (CD) 26)], is a membrane-bound enzyme that serves
functions in metabolism, the immune and endocrine systems, cancer
growth and cell adhesion (17).
DPP4 has been shown to act as a tumor suppressor or
activator by associating with fibroblast activation protein α,
adenosine deaminase, CD45 and C-X-C motif chemokine receptor 4, and
could be considered as a potential therapeutic target in cancers
expressing DPP4 (18,19). In addition, Buffon et al
(20) demonstrated that DPP4
was involved in processes of cervical cancer by regulating cell
migration and adhesion. In the current study, DPP4 was
identified as a differentially methylated gene and DEG associated
with the pathogenesis of cervical cancer and involved in receptor
binding, ameboidal-type cell migration and cell proliferation.
Taken together, these findings indicate that DPP4 is closely
associated with the development of cervical cancer by regulating
cell migration and adhesion.
EDN3 is a member of endothelin family, and
the EDN3 pathway has been shown to be essential for the
proliferation, survival and migration of melanocyte precursor cells
(21,22). Garcia et al (23) demonstrated that EDN3 exhibited
an tumor-angiogenic response in a melanoma mouse model.
Furthermore, EDN3 may be a target of epigenetic
inactivation, affecting the endothelin signalling pathway in human
breast cancer, and hypermethylation of the EDN3 promoter
could lead to gene silencing (24).
In addition, Espinosa et al (25) revealed that the expression of
EDN3 was downregulated in cervical cancer, and Chen et
al (26) revealed that DNA
methylation of EDN3 could be considered a cancer biomarker
in cervical cancer. The current findings were consistent with these
previous reports; EDN3 was identified as differentially
methylated gene and DEG related to cervical cancer, and was
indicated to be involved in ameboidal-type cell migration, MAPK
cascade and cell proliferation. Based on these findings, it may be
speculated that EDN3 is involved in the development of
cervical cancer.
FGF14 is a member of the fibroblast growth
factor (FGF) family, which includes four homologous factors:
FGF11, FGF12, FGF13 and FGF14 (27). Upregulated expression of FGF13
has been demonstrated to mediate the resistance to platinum-based
drugs in cervical cancer cells (28).
Thus, as a homolog of FGF13, it is possible that
FGF14 is involved in cervical cancer development. However,
few studies have reported on the correlation between FGF14
and cervical cancer (28,29). In the current study, using a
comprehensive analysis of microarray data, EDN3 was
identified to be a differentially methylated gene and DEG
associated with cervical cancer. These findings suggest that
FGF14 may be involved in the development of cervical
cancer.
Previously, methylation of TAC1 has been
demonstrated to have a potential correlation with prognosis in
cervical cancer (2). Consistently,
the results of the current study implied that TAC1 was
associated with the pathogenesis of cervical cancer. In addition,
WNT16 is important in oncogenesis, and the WNT signalling
pathway is related to β-catenin signaling and prostate cancer
development (30,31). However, few studies have reported on
the participation of WNT16 in the development of cervical
cancer. In the current study, TAC1 and WNT16 were
identified as differentially methylated genes and DEGs in cervical
cancer. Taken together, it may be speculated that TAC1 and
WNT16 are involved in cervical cancer development.
In summary, the present study identified 91
downregulated and 151 upregulated DEGs in the GSE55940 data set. In
GSE46306, 561 overlapping differentially methylated genes based on
a differential methylation analysis at the CpG site level, CpG
island level and gene level were screened. A total of 5 overlapping
genes (DPP4, EDN3, FGF14, TAC1 and
WNT16) of the 561 overlapping differentially methylated
genes and 242 DEGs were identified, which were downregulated and
hypermethylated in cervical cancer samples. DPP4,
EDN3, FGF14, TAC1 and WNT16 may be
involved in the pathogenesis of cervical cancer. However, the
results must be confirmed by further experiments.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van Ham MA, Bakkers JM, Harbers GK, Quint
WG, Massuger LF and Melchers WJ: Comparison of two commercial
assays for detection of human papillomavirus (HPV) in cervical
scrape specimens: Validation of the Roche AMPLICOR HPV test as a
means to screen for HPV genotypes associated with a higher risk of
cervical disorders. J Clin Microbiol. 43:2662–2667. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sood S, Patel FD, Ghosh S, Arora A,
Dhaliwal LK and Srinivasan R: Epigenetic alteration by DNA
methylation of ESR1, MYOD1 and hTERT gene promoters is useful for
prediction of response in patients of locally advanced invasive
cervical carcinoma treated by chemoradiation. Clin Oncol (R Coll
Radiol). 27:720–727. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clarke MA, Wentzensen N, Mirabello L,
Ghosh A, Wacholder S, Harari A, Lorincz A, Schiffman M and Burk RD:
Human papillomavirus DNA methylation as a potential biomarker for
cervical cancer. Cancer Epidemiol Biomarkers Prev. 21:2125–2137.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kalantari M, Osann K, Calleja-Macias IE,
Kim S, Yan B, Jordan S, Chase DM, Tewari KS and Bernard HU:
Methylation of human papillomavirus 16, 18, 31 and 45 L2 and L1
genes and the cellular DAPK gene: Considerations for use as
biomarkers of the progression of cervical neoplasia. Virology.
448:314–321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nye MD, Hoyo C, Huang Z, Vidal AC, Wang F,
Overcash F, Smith JS, Vasquez B, Hernandez B, Swai B, et al:
Associations between methylation of paternally expressed gene 3
(PEG3), cervical intraepithelial neoplasia and invasive cervical
cancer. PLoS One. 8:e563252013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen CC, Lee KD, Pai MY, Chu PY, Hsu CC,
Chiu CC, Chen LT, Chang JY, Hsiao SH and Leu YW: Changes in DNA
methylation are associated with the development of drug resistance
in cervical cancer cells. Cancer Cell Int. 15:982015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lando M, Fjeldbo CS, Wilting SM, Snoek CB,
Aarnes EK, Forsberg MF, Kristensen GB, Steenbergen RD and Lyng H:
Interplay between promoter methylation and chromosomal loss in gene
silencing at 3p11-p14 in cervical cancer. Epigenetics. 10:970–980.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun NX, Ye C, Zhao Q, Zhang Q, Xu C, Wang
SB, Jin ZJ, Sun SH, Wang F and Li W: Long noncoding RNA-EBIC
promotes tumor cell invasion by binding to EZH2 and repressing
E-cadherin in cervical cancer. PLoS One. 9:e1003402014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ye C, Sun NX, Ma Y, Zhao Q, Zhang Q, Xu C,
Wang SB, Sun SH, Wang F and Li W: MicroRNA-145 contributes to
enhancing radiosensitivity of cervical cancer cells. FEBS Lett.
589:702–709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burris HH, Baccarelli AA, Motta V, Byun
HM, Just AC, Mercado-Garcia A, Schwartz J, Svensson K, Téllez-Rojo
MM and Wright RO: Association between length of gestation and
cervical DNA methylation of PTGER2 and LINE 1-HS. Epigenetics.
9:1083–1091. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang D, Yan L, Hu Q, Sucheston LE, Higgins
MJ, Ambrosone CB, Johnson CS, Smiraglia DJ and Liu S: IMA: An R
package for high-throughput analysis of Illumina's 450K Infinium
methylation data. Bioinformatics. 28:729–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Almouzni G and Cedar H: Maintenance of
Epigenetic Information. Cold Spring Harb Perspect Biol. 8:pii:
a019372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin FF, Wang N, Bi XN, Yu X, Xu XH, Wang
YL, Zhao CQ, Luo B and Wang YK: Serine/threonine kinases 31(STK31)
may be a novel cellular target gene for the HPV16 oncogene E7 with
potential as a DNA hypomethylation biomarker in cervical cancer.
Virol J. 13:602016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi CH, Chung JY, Kim JH, Kim BG and
Hewitt SM: Expression of fibroblast growth factor receptor family
members is associated with prognosis in early stage cervical cancer
patients. J Transl Med. 14:1242016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kotacková L, Baláziová E and Sedo A:
Expression pattern of dipeptidyl peptidase IV activity and/or
structure homologues in cancer. Folia Biol (Praha). 55:77–84.
2009.PubMed/NCBI
|
|
18
|
Havre PA, Abe M, Urasaki Y, Ohnuma K,
Morimoto C and Dang NH: The role of CD26/dipeptidyl peptidase IV in
cancer. Front Biosci. 13:1634–1645. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Busek P, Krepela E, Mares V, Vlasicova K,
Sevcik J and Sedo A: Expression and function of dipeptidyl
peptidase IV and related enzymes in cancer. Adv Exp Med Biol.
575:55–62. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buffon A, Beckenkamp A, Santana DB,
Nascimento J, Paccez J, Zerbini LF, Wink MR and Bruno AN: Abstract
A43: Investigation of dipeptidyl peptidase IV/CD26 role in cervical
cancer cell lines. Mol Cancer Ther. 12:A432013. View Article : Google Scholar
|
|
21
|
Tang L, Su M, Zhang Y, Ip W, Martinka M,
Huang C and Zhou Y: Endothelin-3 is produced by metastatic melanoma
cells and promotes melanoma cell survival. J Cutan Med Surg.
12:64–70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benaduce AP, Batista D, Grilo G, Jorge K,
Cardero D, Milikowski C and Kos L: Novel UV-induced melanoma mouse
model dependent on Endothelin3 signaling. Pigment Cell Melanoma
Res. 27:839–842. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garcia RJ, Ittah A, Mirabal S, Figueroa J,
Lopez L, Glick AB and Kos L: Endothelin 3 induces skin pigmentation
in a keratin-driven inducible mouse model. J Invest Dermatol.
128:131–142. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wiesmann F, Veeck J, Galm O, Hartmann A,
Esteller M, Knüchel R and Dahl E: Frequent loss of endothelin-3
(EDN3) expression due to epigenetic inactivation in human breast
cancer. Breast Cancer Res. 11:R342009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Espinosa AM, Alfaro A, Roman-Basaure E,
Guardado-Estrada M, Palma Í, Serralde C, Medina I, Juárez E,
Bermúdez M, Márquez E, et al: Mitosis is a source of potential
markers for screening and survival and therapeutic targets in
cervical cancer. PLoS One. 8:e559752013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen YC, Huang RL, Huang YK, Liao YP, Su
PH, Wang HC, Chang CC, Lin YW, Yu MH, Chu TY and Lai HC:
Methylomics analysis identifies epigenetically silenced genes and
implies an activation of β-catenin signaling in cervical cancer.
Int J Cancer. 135:117–127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kelleher FC, O'Sullivan H, Smyth E,
McDermott R and Viterbo A: Fibroblast growth factor receptors,
developmental corruption and malignant disease. Carcinogenesis.
34:2198–2205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Okada T, Murata K, Hirose R, Matsuda C,
Komatsu T, Ikekita M, Nakawatari M, Nakayama F, Wakatsuki M, Ohno
T, et al: Upregulated expression of FGF13/FHF2 mediates resistance
to platinum drugs in cervical cancer cells. Sci Rep. 3:28992013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang SS, Smiraglia DJ, Wu YZ, Ghosh S,
Rader JS, Cho KR, Bonfiglio TA, Nayar R, Plass C and Sherman ME:
Identification of novel methylation markers in cervical cancer
using restriction landmark genomic scanning. Cancer Res.
68:2489–2249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ruterbusch JJ, Levin AM, Kittles R,
Rybicki BA and Bock CH: Abstract A66: Admixture and fine mapping in
African Americans identifies a susceptibility locus for prostate
cancer on chromosome 7. Cancer Epidemiol Biomarkers Prev.
21:A662012. View Article : Google Scholar
|