Introduction
Neurofibromatosis type 1 (NF1), which is also known
as von Recklinghausen's disease, is a genetic disorder that
involves autosomal-dominant mutations. The disease is characterized
by skin lesions called café-au-lait spots and cutaneous
neurofibromas (1). The prevalence of
NF1 in general population is ~1 in 3,500 (2). NF1 is associated with mutations in the
neurofibromin 1 (NF1) gene, which generate an increased risk
of variety of tumor types, including benign and malignant tumors
(3). Studies have suggested that
0.1–5.7% of cases of NF1 are complicated with pheochromocytoma
(PHEO) (4). The incidence of PHEO is
2–8 per 1,000,000 adults (4). PHEO
releases catecholamines, which results in a series of clinical
manifestations that include hypertension and metabolic disorders. A
total of 4–25% of patients with NF1 may also present with
gastrointestinal stromal tumors (GIST) (5,6), which are
the most common mesenchymal tumors of the gastrointestinal tract.
GISTs are considered to occur in 20–40 per million inhabitants per
year (7,8), and 20–50% of GISTs are localized in the
small intestine, usually in the jejunum (7,8).
The current study presents a case of NF1 occurring
with PHEO and GIST. To the best of our knowledge, this case is
extremely rare. In addition, a brief literature review is
presented.
Case report
On October 23, 2013, a 56-year-old male was admitted
to the Emergency Department of Ningxia People's Hospital (Yinchuan,
China) for abdominal pain and fever lasting 15 days, concomitant
with cough and polypnea. Upon examination, the patient was found to
have multiple, diffuse soft-tissue lesions measuring 1–5 cm in
diameter located throughout his body, in addition to diffuse
pigmented macules of 2–4 cm in diameter on the skin. A
rough-bordered mass of 10×15 cm diameter was palpable in the upper
left abdomen, and percussion pain was reported in left kidney area.
No optic atrophy, papillary edema or iris Lisch nodules were found
in the fundus oculi. The patient had a history of paroxysmal
hypertension (up to 220/130 mmHg) for >10 years. In addition,
his brother had a history of NF1. Digital radiographic imaging
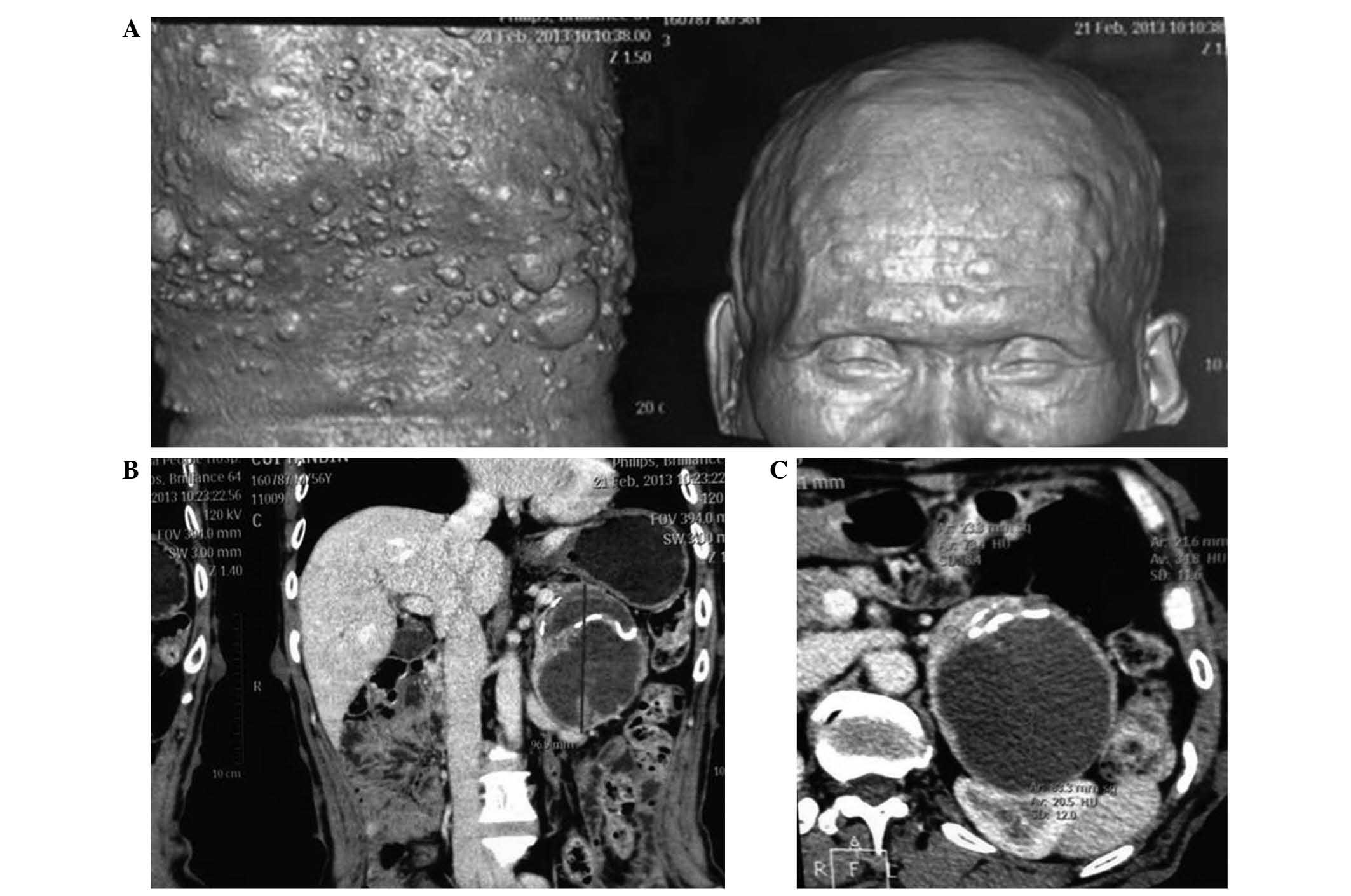
revealed thoracic scoliosis, whilst computed tomography (CT)
imaging (performed using a Diamond Select Brilliance CT 16-slice
scanner; Philips Medical Systems B.V., Eindhoven, The Netherlands)
indicated multiple nodules with soft tissue density, as well as
masses under the scalp and subcutaneous tissue of the abdomen
(Fig. 1A), and an oval-shaped cystic
mass measuring 7.0×7.7×8.9 cm in the left upper abdominal cavity
(Fig. 1B and C).
The patient underwent surgical excision of the
abdominal tumor, small intestine tumor and part of the subcutaneous
nodules for pathological diagnosis. Intraoperative blood pressure
fluctuated significantly and peaked at 300/110 mmHg. Postoperative
blood pressure remained stable at 140/90 mmHg. Routine
anti-infection and intravenous fluid therapy were administered
following surgery. For histopathological examination, specimens
were fixed with 10% formalin, embedded in paraffin and stained with
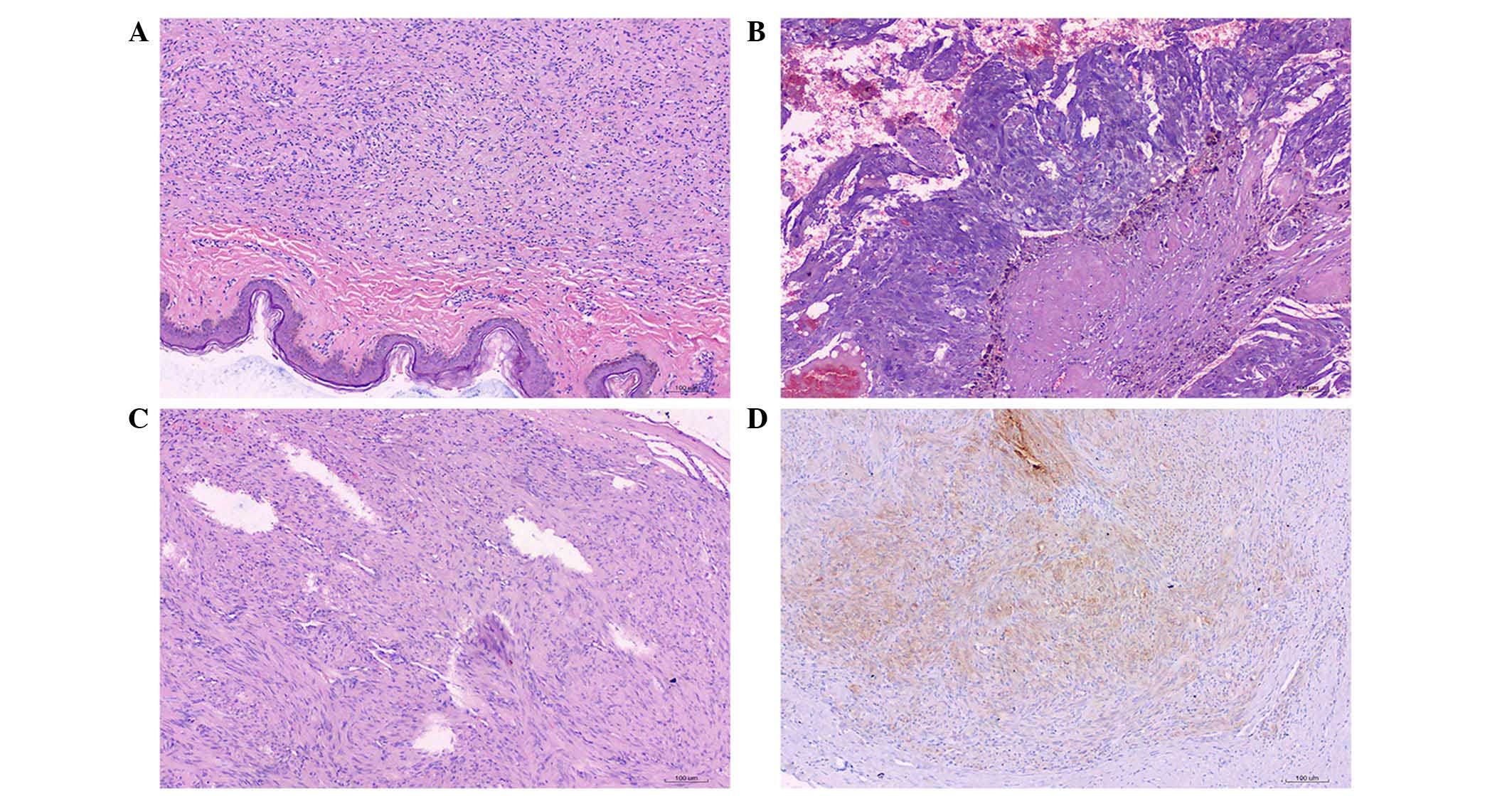
hematoxylin and eosin (H&E). H&E staining of the abdominal
and trunk subcutaneous nodules indicated that the tumor was
composed of loose spindle cells, with little, lightly stained
cytoplasm, and thin, wavy nuclei, and the cells were embedded in a
fiber-like matrix (Fig. 2A).
The resected tumor from the adrenal gland was
grey-red and measured 7.0×7.8×9.0 cm in size. H&E staining of
the resected tumor revealed that the tumor capsule was calcified
and the tumor contained areas of hemorrhage, necrosis and
cholesterol clefts. The tumor cells in areas without hemorrhage and
necrosis were arranged in an organoid pattern, with rich
vascularity; the cells were moderately abnormal. (Fig. 2B). For immunohistochemical analysis,
sections were incubated with antibodies for cluster of
differentiation (CD)117 (polyclonal rabbit anti-human; dilution,
1:50; #A4502), S-100 (polyclonal rabbit anti-human; dilution,
1:4,000; #Z0311), Ki-67 (monoclonal rabbit anti-human; dilution,
1:200; #7240), synaptophysin (polyclonal rabbit anti-human;
dilution, 1:200; #A010), chromogranin A (polyclonal rabbit
anti-human; dilution, 1:500; #A0430), neuron-specific enolase,
cytokeratin (CK)7 (polyclonal rabbit; dilution, 1:500; #A0575) and
carcinoembryonic antigen (polyclonal rabbit anti-human; dilution,
1:500; #M7072) for 24 h at 37°C (all purchased from Dako, Glostrup,
Denmark). The immunohistochemical findings were S-100 (++),
synaptophysin (++), chromogranin A (−), neuron-specific enolase
(−), CK7 (−), carcinoembryonic antigen (−), Ki-67 (−), p53 (−) and
CK18 (−).
The resected small intestine tumor was gray-white
and measured 1.3×1.3×1.0 cm. Upon H&E staining, the spindle
cells in the stomach muscularis were found to be arranged in
bundles, and the nuclei were surrounded by vacuoles (Fig. 2C). Immunohistochemical staining showed
S-100 (−), CD117 (++), CD34 (++), smooth muscle actin (++) and
Ki-67 (−) (Fig. 2D). Combining the
clinical and pathological findings, the patient was diagnosed with
NF1, giant PHEO and small intestinal stromal tumor.
During the one-year postoperative follow-up, the
blood pressure of the patient remained in the normal range, and the
skin nodules softened and reduced in size. No abnormal lesions were
detected in abdominal CT imaging at 6 months post-surgery, and
during a 2-year telephone follow-up, the patient stated that they
had not experienced abnormal blood pressure or other anomalies.
Written informed consent for publication of personal and medical
information were obtained from the patient.
Discussion
The present study conducted a literature search for
relevant articles using Pubmed (January 1950 to October 2014), OVID
(1951 to October 2014), and Web of Science (1900 to October 2014).
To minimize the chance of missing an important study, a manual
search of the references of all articles found in our search was
also performed, including any potentially eligible studies that
were found using Google Scholar. The keywords were as follows:
‘Neurofibromatosis’, ‘pheochromocytoma’ and ‘gastrointestinal
stromal tumors’. As shown in Table I,
in addition to the current patient, only 11 similar cases involving
a coincidence of GIST and PHEO in NF1 have been published worldwide
(9–17). A total of 10 cases reported the
position of PHEO, in which 5 cases occurred in the bilateral
adrenal gland; 11 cases reported the position of GISTs, in which 10
cases were located along the small intestine and 2 case was
malignant; 8 cases reported clinical features, in which 6 patients
had a history of hypertension, 1 patient had melena, 1 had chest
pain and 1 had respiratory insufficiency; 7 cases reported clinical
outcomes, in which 3 patients succumbed to pulmonary embolism,
respiratory insufficiency or intestinal ischemic necrosis,
respectively. NF1 is associated with a defect in the NF1
gene, a tumor suppressor gene located on chromosome 17q11.2.
NF1 encodes neurofibromin, which is responsible for
inhibiting the activity of the p21 Ras protein, and induces a
variety of clinical manifestations (18). Due to a lack of obvious mutation
hotspots, clinical diagnosis of NF1 is still determined based on
the criteria of the National Institutes of Health Consensus
Development Conference, 1987 (19),
which is documented in Table II.
 | Table I.Cases with the coincidence of
gastrointestinal stromal tumors and pheochromocytoma in
neurofibromatosis type 1. |
Table I.
Cases with the coincidence of
gastrointestinal stromal tumors and pheochromocytoma in
neurofibromatosis type 1.
| Author, year | Location | Patient gender/age,
years | Position and size of
tumors | Previous medical and
family history | Clinical
manifestation | Laboratory
findingsa | Immunohistochemical
staining results | Outcome | Ref. |
|---|
| Vlenterie et
al, 2013b | Netherlands | F/59 | Left adrenal gland,
stomach and small intestine | – | – | – | – | Succumbed to
pulmonary embolism | (9) |
| Vlenterie et
al, 2013b | Netherlands | M/55 | Bilateral adrenal
gland, jejunum | – | – | – | – | Survived | (9) |
| Ozcinar et al,
2013 | Turkey | M/48 | Right adrenal gland
(2.2 cm), small intestine (1.5–3.5 cm) | Hypertension | Melena, nodule in
subcutaneous tissue, café au lait spots | VMA, 9.13 (1.4–6.6)
mg/24 h; normetanephrine, 598 (105–354) µg/24 h; metanephrine,
1,315 (140–785) µg/24 h | CD34 (+), c-Kit (+),
CD117 (+), SMA (+), desmin (−), S100 (+), Ki-67 (2%) | NA | (10) |
| Kramer et al,
2007 | Germany | F/63 | Left adrenal gland (5
cm), right adrenal gland (3 cm), ileum (6.5 cm), jejunum (3
cm) | Nodule in
subcutaneous tissue for 13 years | NA | VMA mildly increased;
hormone levels normal | Adrenal gland: S100
(+) GIST: CD34 (+), S100 (−), SMA (−), MIB-1 (2%) | NA | (11) |
| Teramoto et
al, 2007 | Japan | F/60 | Left adrenal gland (3
cm), small intestine (0.5–2 cm) | Hypertension | Headache, nausea | Blood/urine levels of
catecholamines increased | CD34 (+), CD117
(+) | Survived | (12) |
| Teramoto et
al, 2007 | Japan | F/67 | Bilateral adrenal
gland, descending colon (3 cm) | Hashimoto's disease,
intestinal polyp, hypertension | Hypertension | Blood/urine levels
of catecholamines increased | CD34 (+), CD117
(+) | Succumbed to
intestinal ischemic necrosis | (12) |
| Bümming et
al, 2006 | Sweden | M/64 | Left adrenal gland
(6×5 cm), small intestine and mesentery 2–4 cm | Coronary heart
disease, hypertension | Intermittent
hypertension | Adrenaline (urine
level), 13,000 (<101) nmol/24 h, noradrenaline, 15,000 (<551)
nmol/24 h; OMC, 72 (<7) nmol/24 h; VMA, 650 (<34) nmol/24 h;
plasma CgA, 400 (<45) U/I | Ki-67 (<1%),
S100 (+), SMA (−), CD117 (+) | NA | (13) |
| Lou et al,
2006 | China | F/56 | Right adrenal gland
(4×3×2.5 cm), jejunum (3–5.5 cm) | Nodule in
subcutaneous tissue for 50 years, hypertension | Abdominal mass | NA | S100 (+), CD117
(+), CD34 (+) vimentin (+), NSE (+), CgA (+), Syn (+), SMA (−), Bcl
(−), KP-1 (−), CK (−) | Survived | (14) |
| Jeong et al,
2005b | Korea | – | – | – | – | – | S100 (+), CD117
(+) | – | (15) |
| Rizzo et al,
2000b | Italy | M/60 | Bilateral adrenal
gland, duodenum, jejunum | – | – | – | – | – | (16) |
| Sakaguchi et
al 1996 | Japan | M/48 | Bilateral adrenal
gland, gastrointestinal tract (malignant) | NA | Chest pain,
paroxysmal hypertension, respiratory insufficiency | NA | NA | Succumbed to
respiratory insufficiency | (17) |
| Table II.Diagnostic criteria for NF1. |
Table II.
Diagnostic criteria for NF1.
| No. | Description |
|---|
| 1 | Six or more café
au lait spots measuring ≥5 mm in diameter in prepubertal
individuals or ≥15 mm in diameter in postpubertal individuals |
| 2 | Two or more
neurofibromas of any type, or one plexiform neurofibroma |
| 3 | Freckling in the
axillary or inguinal regions |
| 4 | Optic glioma |
| 5 | Two or more Lisch
nodules (iris hamartomas) |
| 6 | A distinctive
osseous lesion, such as sphenoid dysplasia or thinning of long bone
cortex, with or without pseudoarthrosis |
| 7 | A first degree
relative (parent, sibling or offspring) with NF1 based on the above
criteria |
NF1 has a wide range of clinical characteristics
(2). The presence of multiple café
au lait spots and peripheral nerve neurofibromas are prominent
features. NF1 is also characterized by several developmental
abnormalities, including an increased frequency of benign and
malignant tumors. A descriptive analysis based on the international
database of the National Neurofibromatosis Foundation (20) suggested that 4.9% of NF1 cases
(n=72/1,479) are complicated by other system tumors. Other studies
have suggested that 0.1–5.7% of NF1 patients have PHEO (4), whilst 4–25% of patients may have NF1
with GIST (6,7). Thus, the occurrence of PHEO and GIST is
rare in patients with NF1.
In a study by Zinnamosca et al (21) of 48 patients with NF-1-related PHEO
(NF1-PHEO) and an average age of 39 years, ~85% of patients had
single adrenal tumors, whilst 15% had bilateral adrenal tumors. In
addition, 57% of patients exhibited the clinical features of high
catecholamine concentration, including headache, heart
palpitations, sweating and hypertension. In previous reports of
clinical cases, paroxysmal hypertension was observed in the
majority of NF1-PHEO patients; these results suggest that the
periodic evaluation of NF1 patients with hypertension for the
presence of PHEO is necessary (21–24).
The development of PHEO may be associated with the
type of NF1 expression. Gutmann et al (25) demonstrated a lack of neurofibromin
expression in PHEO from 6 patients with NF1, supporting the idea
that neurofibromin may participate in the pathogenesis of NF1-PHEO.
Additionally, reduced or absent NF1 gene expression was
documented in a proportion of PHEO patients without NF1.
Furthermore, the majority of PHEO cases expressed predominantly the
type 1 NF1 isoform (75%), as opposed to the adrenal glands, which
express predominantly type 2 NF1 (60%) (25).
The incidence of GIST accounts for only 0.1–3.0% of
all gastrointestinal tumors (26);
however, 11–25% patients with NF1 also have gastrointestinal
carcinoid tumors (27). Although 60%
of GISTs occur in the stomach whilst only 20–30% occur in the small
intestine (28), NF1-related GISTs
(NF1-GIST) tend to be located in the small intestine (28). The pathogenesis of NF1-GIST is
different to that of sporadic GIST. The latter, which is widely
considered to involve specific KIT (CD117) or platelet-derived
growth factor receptor α polypeptide (PDFGRA) signaling-driven
tumors, is characterized by frequent allelic losses of 1p, 14q and
22q, and mutations of the KIT or PDGFRA genes
(6,29). By contrast, mutations in KIT
and PDGFRA are extremely rare events in NF1-GIST (29). Rather, cell proliferation in NF-1 GIST
is associated with the activation of the Ras-mitogen-activated
protein kinase pathway, which may be activated via KIT or
PDGFRA in association with the inactivation of the
NF1 gene. Additionally, loss of heterozygosity at 14q and
22q may contribute to the relatively early phase of tumor
development of NF1-GIST (30). The
diagnosis of GIST primarily depends on the histopathological and
immunohistochemical phenotype. One of the main characteristic of
GIST is positivity for CD117 and CD34 (31,32). In
the majority of the present patients with NF1-GIST, CD117 and CD34
were positive, which is conducive to confirming a diagnosis.
Surgical removal is the only definitive therapy for
PHEO (33). However, it is important
to note that patients who experience an inconspicuous
intraoperative blood pressure drop or rapid recovery of previously
low blood pressure during tumor excision have the possibility for
residual tumor or metastases (33).
Thus, careful monitoring of blood pressure should be conducted
during surgery. In addition, surgical excision is the standard
approach for GIST of >2 cm in diameter or with nuclear
pleomorphism as a histological feature (34). Treatment should be supplemented with
imatinib mesylate for suppression of CD117 receptors and prevention
of tumor recurrence or metastasis (35).
In conclusion, the current study reiterates that
cases of NF1 occurring with PHEO and GIST are rare. The clinical
symptoms of these cases varied; however, paroxysmal hypertension
occurred in the majority of patients, and GISTs were largely
located in the small intestine. The present study aims to offer
guidance in the diagnosis and treatment of similar cases.
References
|
1
|
Riccardi VM: Von Recklinghausen
neurofibromatosis. N Engl J Med. 305:1617–1627. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman JM: Epidemiology of
neurofibromatosis type 1. Am J Med Genet. 89:1–6. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gutmann DH, Aylsworth A, Carey JC, Korf B,
Marks J, Pyeritz RE, Rubenstein A and Viskochil D: The diagnostic
evaluation and multidisciplinary management of neurofibromatosis 1
and neurofibromatosis 2. Jama. 278:51–57. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walther MM, Herring J, Enquist E, Keiser
HR and Linehan WM: von Recklinghausen's disease and
pheochromocytomas. J Urol. 162:1582–1586. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fung MM, Viveros OH and O'Connor DT:
Diseases of the adrenal medulla. Acta Physiol. 192:325–335. 2008.
View Article : Google Scholar
|
|
6
|
Miettinen M, Fetsch JF, Sobin LH and
Lasota J: Gastrointestinal stromal tumors in patients with
neurofibromatosis 1: A clinicopathologic and molecular genetic
study of 45 cases. Am J Surg Pathol. 30:90–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yantiss RK, Rosenberg AE, Sarran L, Besmer
P and Antonescu CR: Multiple gastrointestinal stromal tumors in
type I neurofibromatosis: A pathologic and molecular study. Mod
Pathol. 18:475–484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cichoz-Lach H, Kasztelan-Szczerbińska B
and Słomka M: Gastrointestinal stromal tumors: Epidemiology,
clinical picture, diagnosis, prognosis and treatment. Pol Arch Med
Wewn. 118:216–221. 2008.PubMed/NCBI
|
|
9
|
Vlenterie M, Flucke U, Hofbauer LC,
Timmers HJ, Gastmeier J, Aust DE, van der Graaf WT, Wesseling P,
Eisenhofer G and Lenders JW: Pheochromocytoma and gastrointestinal
stromal tumors in patients with neurofibromatosis type I. Am J Med.
126:174–180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ozcinar B, Aksakal N, Agcaoglu O, Tukenmez
M, Ozemir IA, Barbaros U, Colak N and Erbil Y: Multiple
gastrointestinal stromal tumors and pheochromocytoma in a patient
with von Recklinghausen's disease. Int J Surg Case Rep. 4:216–218.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kramer K, Hasel C, Aschoff AJ, Henne-Bruns
D and Wuerl P: Multiple gastrointestinal stromal tumors and
bilateral pheochromocytoma in neurofibromatosis. World J
Gastroenterol. 13:3384–3387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Teramoto S, Ota T, Maniwa A, Matsui T,
Itaya N, Aoyagi K, Kusanagi H and Narita M: Two von
Recklinghausen's disease cases with pheochromocytomas and
gastrointestinal stromal tumors (GIST) in combination. Int J Urol.
14:73–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bümming P, Nilsson B, Sörensen J, Nilsson
O and Ahlman H: Use of 2-tracer PET to diagnose gastrointestinal
stromal tumour and pheochromocytoma in patients with Carney triad
and neurofibromatosis type 1. Scand J Gastroenterol. 41:626–630.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo ZM, Jin XL and Teng HH:
Neurofibromatosis type I complicating interstitialoma and
pheochromocytoma: A case of report. Chin J Diag Pathol. 36:140–141.
2006.
|
|
15
|
Jeong CY, Hong SC, Lee YJ, Jung EJ, Choi
SK, Joo YT, Ha WS, Park ST and Lee JS: Multiple duodeno-jejunal
GIST associated with pheochromocytoma in patients with von
Recklinghausen disease. J Korean Surg Soc. 69:74–78. 2005.(In
Korean).
|
|
16
|
Rizzo S, Bonomo S, Moser A, Bottura D,
Castellini C, Mazzola F, Lauro E, Vicenzi L, Betresini B, Angeli G,
et al: Bilateral pheochromocytoma associated with duodeno-jejunal
GIST in patient with von Recklinghausen disease: Report of a
clinical case. Chir Ital. 53:243–246. 2001.(In Italian). PubMed/NCBI
|
|
17
|
Sakaguchi N, Sano K, Ito M, Baba T,
Fukuzawa M and Hotchi M: A case of von Recklinghausen's disease
with bilateral pheochromocytoma-malignant peripheral nerve sheath
tumors of the adrenal and gastrointestinal autonomic nerve tumors.
Am J Surg Pathol. 20:889–897. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oguzkan S, Terzi YK, Cinbis M, Anlar B,
Aysun S and Ayter S: Molecular genetic analyses in
neurofibromatosis type 1 patients with tumors. Cancer Genet
Cytogenet. 165:167–171. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Neurofibromatosis. Conference statement.
National Institutes of Health Consensus Development Conference.
Arch Neurol. 45:575–578. 1988.PubMed/NCBI
|
|
20
|
Friedman J and Birch PH: Type 1
neurofibromatosis: A descriptive analysis of the disorder in 1,728
patients. Am J Med Genet. 70:138–143. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zinnamosca L, Petramala L, Cotesta D,
Marinelli C, Schina M, Cianci R, Giustini S, Sciomer S, Anastasi E,
Calvieri S, et al: Neurofibromatosis type 1 (NF1) and
pheochromocytoma: Prevalence, clinical and cardiovascular aspects.
Arch Dermatol Res. 303:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Opocher G, Conton P, Schiavi F, Macino B
and Mantero F: Pheochromocytoma in von Hippel-Lindau disease and
neurofibromatosis type 1. Fam Cancer. 4:13–16. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Erem C, Onder Ersöz H, Ukinç K,
Hacihasanoglu A, Alhan E, Cobanoğlu U, Koçak M and Erdöl H:
Neurofibromatosis type 1 associated with pheochromocytoma: A case
report and a review of the literature. J Endocrinol Invest.
30:59–64. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
March Rocchietti M: Type 1
neurofibromatosis and pheochromocytoma: Focus on hypertension. J
Neurosci Rural Pract. 3:107–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gutmann DH, Geist RT, Rose K, Wallin G and
Moley JF: Loss of neurofibromatosis type I (NFI) gene expression in
pheochromocytomas from patients without NFI. Genes Chromosomes
Cancer. 13:104–109. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Singer S, Rubin BP, Lux ML, Chen CJ,
Demetri GD, Fletcher CD and Fletcher JA: Prognostic value of KIT
mutation type, mitotic activity and histologic subtype in
gastrointestinal stromal tumors. J Clin Oncol. 20:3898–3905. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pinsk I, Dukhno O, Ovnat A and Levy I:
Gastrointestinal complications of von Recklinghausen's disease: Two
case reports and a review of the literature. Scand J Gastroenterol.
38:1275–1278. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Caterino S, Lorenzon L, Petrucciani N,
Iannicelli E, Pilozzi E, Romiti A, Cavallini M and Ziparo V:
Gastrointestinal stromal tumors: Correlation between symptoms at
presentation, tumor location and prognostic factors in 47
consecutive patients. World J Surg Oncol. 9:132011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Maertens O, Prenen H, Debiec-Rychter M,
Wozniak A, Sciot R, Pauwels P, De Wever I, Vermeesch JR, de Raedt
T, De Paepe A, et al: Molecular pathogenesis of multiple
gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet.
15:1015–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamamoto H, Tobo T, Nakamori M, Imamura M,
Kojima A, Oda Y, Nakamura N, Takahira T, Yao T and Tsuneyoshi M:
Neurofibromatosis type 1-related gastrointestinal stromal tumors: A
special reference to loss of heterozygosity at 14q and 22q. J
Cancer Res Clin Oncol. 35:791–798. 2009. View Article : Google Scholar
|
|
31
|
Novelli M, Rossi S, Rodriguez-Justo M,
Taniere P, Seddon B, Toffolatti L, Sartor C, Hogendoorn PC, Sciot
R, Van Glabbeke M, et al: DOG1 and CD117 are the antibodies of
choice in the diagnosis of gastrointestinal stromal tumours.
Histopathology. 57:259–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miettinen M, Majidi M and Lasota J:
Pathology and diagnostic criteria of gastrointestinal stromal
tumors (GISTs): A review. Eur J Cancer. 38:S39–S51. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bruynzeel H, Feelders RA, Groenland THN,
van den Meiracker AH, van Eijck CH, Lange JF, de Herder WW and
Kazemier G: Risk factors for hemodynamic instability during surgery
for pheochromocytoma. J Clin Endocrinol Metab. 95:678–685. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
ESMO/European Sarcoma Network Working
Group: Gastrointestinal stromal tumors: ESMO Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol.
23(Suppl 7): 23. vii49–vii55. 2012.PubMed/NCBI
|
|
35
|
Shinomura Y, Kinoshita K, Tsutsui S and
Hirota S: Pathophysiology, diagnosis and treatment of
gastrointestinal stromal tumors. J Gastroenterol. 40:775–780. 2005.
View Article : Google Scholar : PubMed/NCBI
|