Introduction
The term autophagy (literally ‘self-eating’)
indicates a number of distinct cellular processes, which together
share the outcome of the degradation of intracellular components by
the lysosome, and are critical for maintaining normal cellular
homeostasis (1,2). The role of autophagy in regulating
either cancer cell death or survival remains highly controversial.
Due to the tumor suppressive and tumor promoting properties, at a
first glance, autophagy may appear paradoxical. Indeed, the
knockdown of genes involved in the autophagic process can either
enhance or prevent cancer cell death (3). Acute leukemia (AL) is a malignant
neoplastic hematological disorder, and the role of autophagy in AL
is currently a hot topic. Since the 1990s, studies have highlighted
the occurrence of autophagy in AL cells. Reviewing the ambiguous
importance of autophagy in either promoting or suppressing AL cell
growth and survival, Evangelisti et al (4) indicated that autophagy may represent a
distinct mechanism of cell death, referred to as autophagic cell
death (ACD), which also occurs in established tumors, including AL
(5–8).
In addition, autophagy can promote necroptosis, a non-apoptotic
form of regulated cell death (9–10).
According to these studies, ACD may be a means for treating this
type of hematological malignancy and for enhancing the activity of
current therapies. However, it is first necessary to establish the
factors that control ACD.
There are several signal channels associated with
autophagy, including mammalian target of rapamycin and Beclin1. The
complex that is composed of Beclin1 and B cell lymphoma-2 (Bcl-2)
is the major regulating factor in the Beclin1 channel (11–13).
Beclin1 and Bcl-2 have antagonistic roles; Beclin1 can promote
autophagy, but Bcl-2 can inhibit autophagy. A recent study
indicated that ACD was controlled by caspase-10 through regulating
the balance of Beclin1 and Bcl-2 in multiple myeloma (14). Caspase-10 belongs to the caspase
family, and its paralogue, caspase8, initiates a type of programmed
cell death known as apoptosis (15).
However, Lamy et al (14)
showed that caspase-10 is an important factor in maintaining a
balanced level of autophagy; when caspase-10 was inhibited, ACD of
myeloma cell was caused by the excessive activation of Beclin1.
The study by Liu et al (16) mentioned that common chemotherapy
drugs, including vincristine (VCR), arsenic trioxide and adriamycin
(ADM), may cause the appearance of autophagosomes in AL cells by
causing the increased expression of high mobility group box-1
protein. In addition, a literature review of autophagy in leukemia
cells revealed that no other study had reported that caspase-10 was
associated with autophagy in AL cells (17,18).
Therefore, the present study considered whether ACD could be
controlled by caspase-10 in AL, as in multiple myeloma. In the
present study, the rate of survival of AL cells treated with
chemotherapy drugs combined with a caspase-10 inhibitor was
compared with AL cells treated with chemotherapy drugs alone.
Methods such as flow cytometry and western blotting were used to
investigate whether caspase-10 is associated with apoptosis or
autophagy in this context.
Materials and methods
Reagents
The antibodies to caspase-3 were obtained from Cell
Signaling Technology (Danvers, MA, USA). The caspase-10 antibody
was obtained from Abcam (Shanghai, China). Cytosine arabinoside was
purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-caspase 3
antibody (monoclonal, rabbit; 1:1,000 dilution; catalog no. 9662)
was purchased from Cell Signaling Technology, anti-caspase-10
antibody (monoclonal, rabbit; 1:1,000 dilution; catalog no.
ab177475) was purchased from Abcam and anti-GAPDH antibody
(control; polyclonal, rabbit; 1:5,000 dilution; catalog no. AP0063)
was purchased from Bioworld Technology Inc. (St. Louis Park, MN,
USA). Goat anti-rabbit immunoglobulin G-horse radish peroxidase
conjugated secondary antibody (1:500-1:5,000 dilution; catalog no.
BS13278) was purchased from Bioworld Technology Inc. VCR, arsenic
trioxide, ADM, the caspase-10 inhibitor (z-aevd-fmk) and
3-methyladenine (3-MA) were purchased from Sigma-Aldrich.
Cell culture
The human leukemia cell lines, HL-60 (derived from
an acute myeloid leukemia) and Jurkat (derived from an acute
lymphoid leukemia), were obtained from the American Type Culture
Collection (Manassas, VA, USA) and cultured in Iscove's modified
Dulbecco's medium or RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), respectively, with 10%
heat-inactivated fetal bovine serum, 2 mM glutamine and an
antibiotic-antimycotic mix (containing 10,000 U/ml penicillin,
10,000 µg/ml streptomycin and 25 µg/ml amphotericin B), in a
humidified incubator with 5% CO2.
Cell viability assay
Cells were plated at a density of 5×104
cells/well in 96-well plates in 100 µl medium. The chemotherapy
drugs used for the treatment were VCR (1 µg/ml), ADM (1 µg/ml),
cytosine arabinoside (Ara-c; 0.2 µM) and arsenic trioxide
(AS2O3; 2.5 µM). Following treatment for 24
h, cell viability was evaluated using Cell Counting Kit-8 (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan), according to the
manufacturer's instructions. In parallel, an analysis of cell
viability by trypan blue (Sigma-Aldrich; catalog no. 302643) MSDS
exclusion assay was performed according the manufacturer's
instructions, which yielded similar results (data not shown).
Assessment of apoptosis by flow cytometric analysis using Annexin
V-phycoerythrin (PE; Sigma-Aldrich) was used in conjunction with a
vital dye, 7-amino actinomycin D (7-AAD), to distinguish apoptotic
(Annexin V-PE positive, 7-AAD negative) from necrotic (Annexin V-PE
positive, 7-AAD positive) cells. After treatment, cells were
trypsinized, collected and resuspended in 40 µl binding buffer with
2 µl Annexin V-PE. Cells were incubated for 15 min in the dark at
room temperature. Subsequent to incubation, 160 µl binding buffer
and 2 µl 7-AAD were added. Cells were then incubated for 5 min and
additional 200 µl binding buffer was added. Prior to analysis, the
cells were filtered through a cell strainer cap fitted to a
polystyrene, round bottomed, flow cytometric tube. Data were
collected and analyzed by a FACS Calibur flow cytometer with
CellQuest Pro software version 5.1 (BD Biosciences, Franklin Lakes,
NJ, USA), with a total of 10,000 events per sample. Fluorescence
was measured and the percentage of viable, early apoptotic, late
apoptotic and necrotic cells was determined.
Western blot analysis
Cells were lysed in 20 mM Tris (pH 7.5), 150 mM
NaCl, 1 mM ethylenediaminetetraacetic acid and 1% Tergitol-type
NP-40 supplemented with a protease inhibitor (Complete Mini
EDTA-free; Roche Diagnostics, Indianapolis, IN, USA) and
phosphatase inhibitor mixture II (Sigma-Aldrich). In all, 30 mg of
protein per lane was run on a denaturing 10% sodium dodecyl
sulfate-polyacrylamide gel and subsequently transferred to
polyvinylidene fluoride membranes via semidry transfer. After
blocking the membrane at room temperature for 3 h, the membrane was
incubated overnight at 4°C with anti-caspase-3 and −10 antibodies.
After incubation with peroxidase-conjugated secondary antibodies
for 1 h at 25°C, the signals were visualized using enhanced
chemiluminescence (Pierce; Thermo Fisher Scientific, Inc.).
Statistical analysis
Results were expressed as mean ± standard deviation
(µ ± SD). The significance of differences was calculated by one-way
analysis of variance, and the threshold for indicating a
significant difference was defined as P<0.05. SPSS 20.0 (IBM
SPSS, Armonk, NY, USA) was used for all statistical analyses.
Results
Survival rate of HL-60 and Jurkat
cells treated with chemotherapy drugs or chemotherapy drugs
combined with a caspase-10 inhibitor
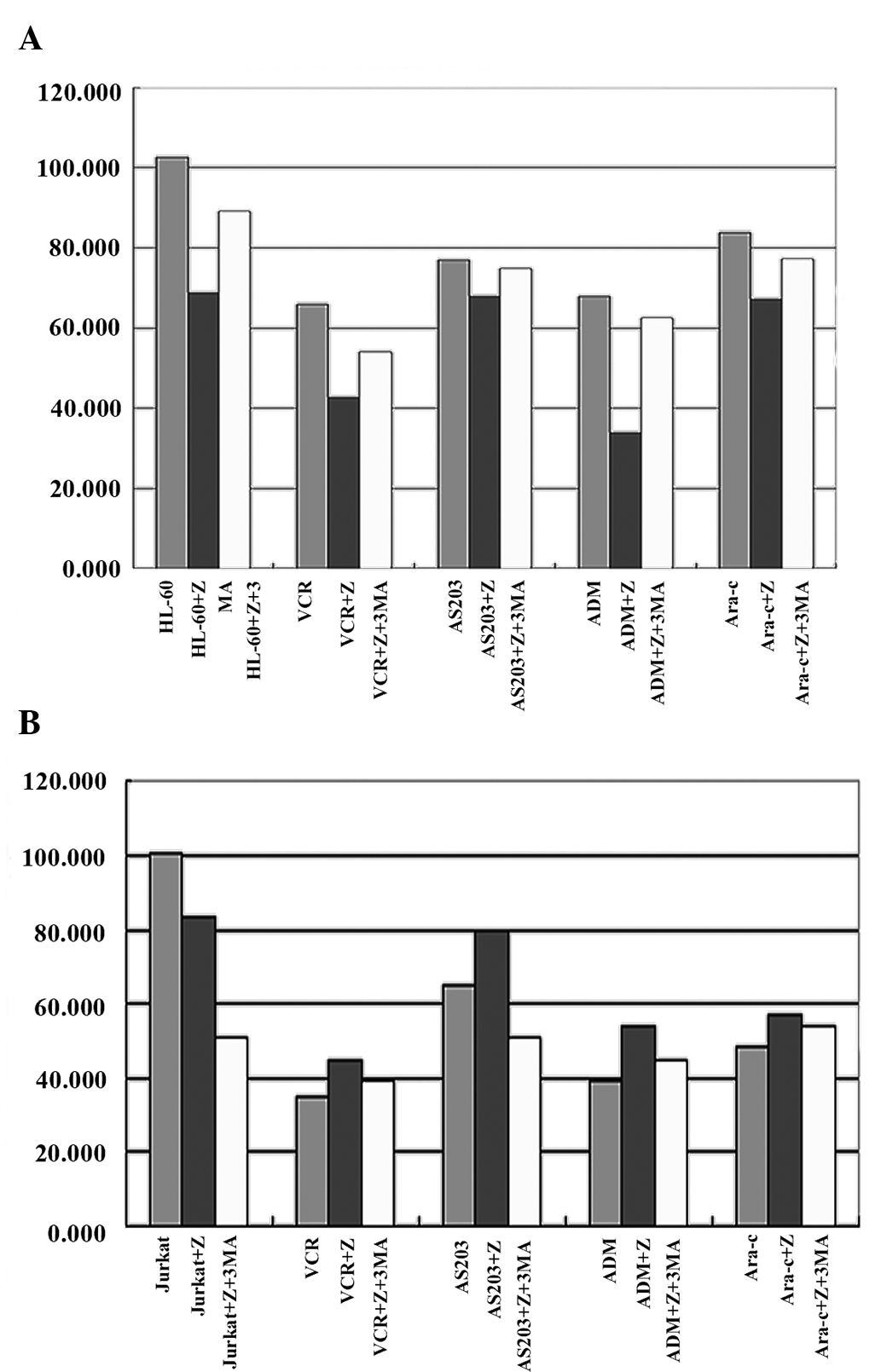
The survival rates of HL-60 cells treated with the
following chemotherapy drugs alone were:
AS2O3, 76.92±8.44; ADM, 67.92±4.39; VCR,
65.86±20.25; and Ara-c, 83.81±15.11. The survival rate of HL-60
cells treated with chemotherapy drugs combined with a caspase-10
inhibitor (z-aevd-fmk) was lower compared with the group treated
with only chemotherapy drugs. The survival rates of HL-60 cells
treated with chemotherapy drugs combined with the caspase-10
inhibitor were: AS2O3, 67.83±4.81; ADM,
33.89±2.48; VCR, 42.70±3.70; and Ara-c, 69.11+1.11. The differences
were significant. The survival rate of the HL-60 cells was also
significantly decreased compared with the control when they were
treated with the caspase-10 inhibitor alone (P=0.023; Table I; Fig.
1A). The memebers of the caspase family are known to be
important priming factors for apoptosis. However, the change in the
survival rate of HL-60 cells appears to suggest that caspase-10 may
not be associated with apoptosis in HL-60 cells.
 | Figure 1.Survival rates in HL-60 and Jurkat
cells when treated with a caspase-10 inhibitor (z-aevd-fmk), a
caspase-10 inhibitor (z-aevd-fmk) with 3-MA, a chemotherapy drug, a
chemotherapy drug combined with a caspase-10 inhibitor (z-aevd-fmk)
and a chemotherapy drug combined with a caspase-10 inhibitor
(z-aevd-fmk) and 3-MA: (A) Survival rate in HL-60 cells when
treated with a caspase-10 inhibitor (z-aevd-fmk), a caspase-10
inhibitor (z-aevd-fmk) with 3-MA, a chemotherapy drug, a
chemotherapy drug combined with a caspase-10 inhibitor (z-aevd-fmk)
and a chemotherapy drug combined with a caspase-10 inhibitor
(z-aevd-fmk) and 3-MA; (B) Survival rate in Jurkat cells when
treated with a caspase-10 inhibitor (z-aevd-fmk), a caspase-10
inhibitor (z-aevd-fmk) with 3-MA, a chemotherapy drug, a
chemotherapy drug combined with a caspase-10 inhibitor (z-aevd-fmk)
and a chemotherapy drug combined with a caspase-10 inhibitor
(z-aevd-fmk) and 3-MA. Z, caspase-10 inhibitor (z-aevd-fmk); 3-MA,
3-methyladenine; VCR, vincristine; AS2O3,
arsenic trioxide; ADM, adriamycin; Ara-c, cytosine arabinoside. |
 | Table I.Survival and apoptotic rates of HL-60
and Jurkat cells treated with chemotherapy drugs or chemotherapy
drug combined with z-aevd-fmk for 24 h. |
Table I.
Survival and apoptotic rates of HL-60
and Jurkat cells treated with chemotherapy drugs or chemotherapy
drug combined with z-aevd-fmk for 24 h.
|
| HL-60 cell | Jurkat cell |
|---|
|
|
|
|
|---|
| Treatment | Survival (%) | Apoptosis (%) | Survival (%) | Apoptosis (%) |
|---|
| Control |
102.55±9.46a |
0.51±0.11b |
100.62±8.73a |
0.62±0.12a |
|
AS2O3 |
76.92±8.44a |
4.91±1.50b |
64.65±11.16a |
5.11±2.90a |
| ADM |
67.92±4.39a |
10.02±4.20b |
39.34±1.80a |
35.40±7.80a |
| VCR |
65.86±20.25a |
12.63±5.90b |
34.61±1.26a |
45.42±9.80a |
| Ara-c |
83.81±15.11a |
2.12±1.20b |
48.50±0.75a |
20.20±3.20a |
| Z+blank |
68.87±4.31a |
0.50±0.30b |
83.77±3.87a |
0.53±0.10b |
|
Z+AS2O3 |
67.83±4.81a |
3.61±1.80b |
79.27±1.83a |
1.10±0.90a |
| Z+ADM |
33.89±2.48a |
9.01±3.40b |
54.01±2.85a |
12.21±5.60a |
| Z+VCR |
42.70±3.70a |
10.50±5.40b |
44.86±1.05a |
14.30±5.40a |
| Z+Ara-c |
69.11±1.11a |
3.62±1.10b |
57.23±0.49a |
6.72±2.10a |
The survival rates of Jurkat cells treated with only
the chemotherapy drugs were: AS2O3,
64.65±11.16; ADM, 39.34±1.80; VCR, 34.61±1.26; and Ara-c,
48.50±0.75. The survival rate of Jurkat cells treated with the
chemotherapy drugs combined with the caspase-10 inhibitor was
increased compared with the group treated with only the
chemotherapy drugs. The survival rates of Jurkat cells treated with
the chemotherapy drugs combined with the caspase-10 inhibitor were:
AS2O3, 79.27±1.83; ADM, 54.01±2.85; VCR,
44.86±1.05; Ara-c, 57.23±0.49. The differences were significant.
However, the survival rate of Jurkat cells also significantly
decreased compared with the control when it was treated with only
the caspase-10 inhibitor (P=0.019; Table
I; Fig. 1B). The change in the
survival rate of Jurkat cells treated with chemotherapy drugs
combined with caspase-10 inhibitor was different from HL-60 cells,
and the result showed that caspase-10 was associated with the
apoptosis in Jurkat cells. However, the change in the survival rate
of Jurkat cells treated with only the caspase-10 inhibitor was
similar to the survival rate of HL-60 cells.
Rate of apoptosis in HL-60 and Jurkat
cells treated with chemotherapy drugs or chemotherapy drugs
combined with a caspase-10 inhibitor
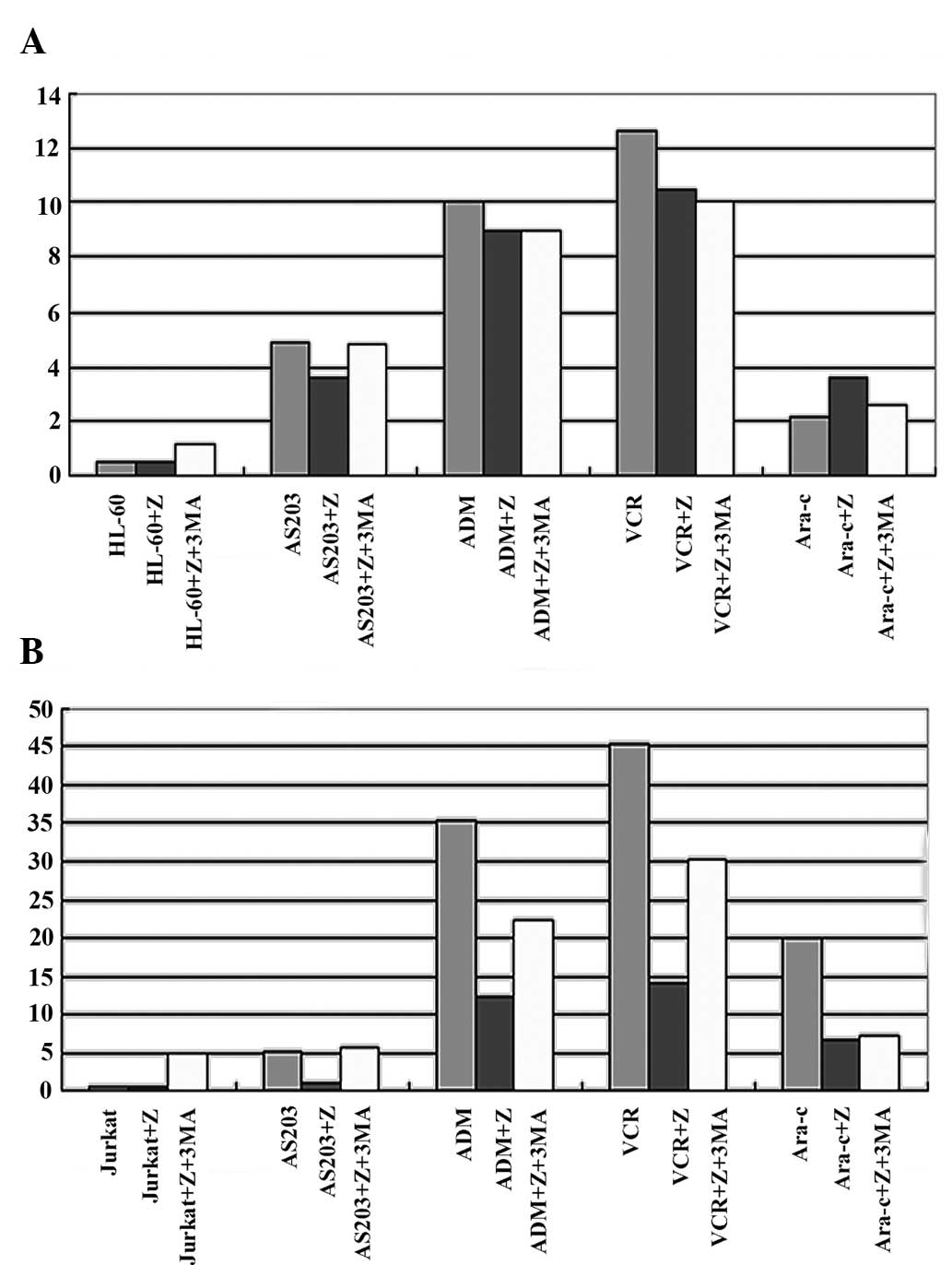
According to the change in survival rate of HL-60
treated by chemotherapy drugs combined with caspase-10 inhibitor,
the present study examined whether the result was associated with
the change in the rate of apoptosis. Thus, the rate of apoptosis in
HL-60 cell treated in various ways was tested. The rates of
apoptosis in HL-60 cells treated with chemotherapy drugs for 24 h
were: AS2O3, 4.9±1.5; ADM, 10.0±4.2; VCR,
12.6±5.9; Ara-c, 2.1±1.2. However, there was no significant
difference in the rate of apoptosis between the group treated with
chemotherapy drugs combined with caspase-10 inhibitor and the group
treated with only the chemotherapy drugs. In addition, there was no
significant difference in the rate of apoptosis in HL-60 cells
treated with only the caspase-10 inhibitor compared with the
control (P=0.231; Table I; Fig. 2A). This result showed that the change
in survival rate could not be explained by apoptosis.
 | Figure 2.Rate of apoptosis in HL-60 and Jurkat
cells when treated with a caspase-10 inhibitor (z-aevd-fmk), a
caspase-10 inhibitor (z-aevd-fmk) with 3-MA, a chemotherapy drug, a
chemotherapy drug combined with a caspase-10 inhibitor (z-aevd-fmk)
and a chemotherapy drug combined with a caspase-10 inhibitor
(z-aevd-fmk) and 3-MA: (A) Rate of apoptosis in HL-60 cells when
treated with a caspase-10 inhibitor (z-aevd-fmk), a caspase-10
inhibitor (z-aevd-fmk) with 3-MA, a chemotherapy drug, a
chemotherapy drug combined with a caspase-10 inhibitor (z-aevd-fmk)
and a chemotherapy drug combined with a caspase-10 inhibitor
(z-aevd-fmk) and 3-MA; (B) Rate of apoptosis in Jurkat cells when
treated with a caspase-10 inhibitor (z-aevd-fmk), a caspase-10
inhibitor (z-aevd-fmk) with 3-MA, a chemotherapy drug, a
chemotherapy drug combined with a caspase-10 inhibitor (z-aevd-fmk)
and a chemotherapy drug combined with a caspase-10 inhibitor
(z-aevd-fmk) and 3-MA. Z, caspase-10 inhibitor (z-aevd-fmk); 3-MA,
3-methyladenine; AS2O3, arsenic trioxide;
ADM, adriamycin; VCR, vincristine; Ara-c, cytosine arabinoside. |
The change in the rate of apoptosis was different
between the Jurkat cells and the HL-60 cells. The rates of
apoptosis in Jurkat cells treated with the chemotherapy drugs for
24 h were: AS2O3, 5.1±2.9; ADM, 35.4±7.8;
VCR, 45.4±9.8; Ara-c, 20.2±3.2. The rate of apoptosis decreased
significantly compared with the control in the Jurkat cells treated
with chemotherapy drugs combined with caspase-10 inhibitor
(P=0.021; Table I, Fig. 2B). However, there was no significant
difference in the rate of apoptosis in the Jurkat cells treated
with only the caspase-10 inhibitor (P=0.114; Table I; Fig.
2B). The change in the rate of apoptosis was also different
between the Jurkat cells treated by chemotherapy drugs combined
with caspase-10 inhibitor and only the caspase-10 inhibitor.
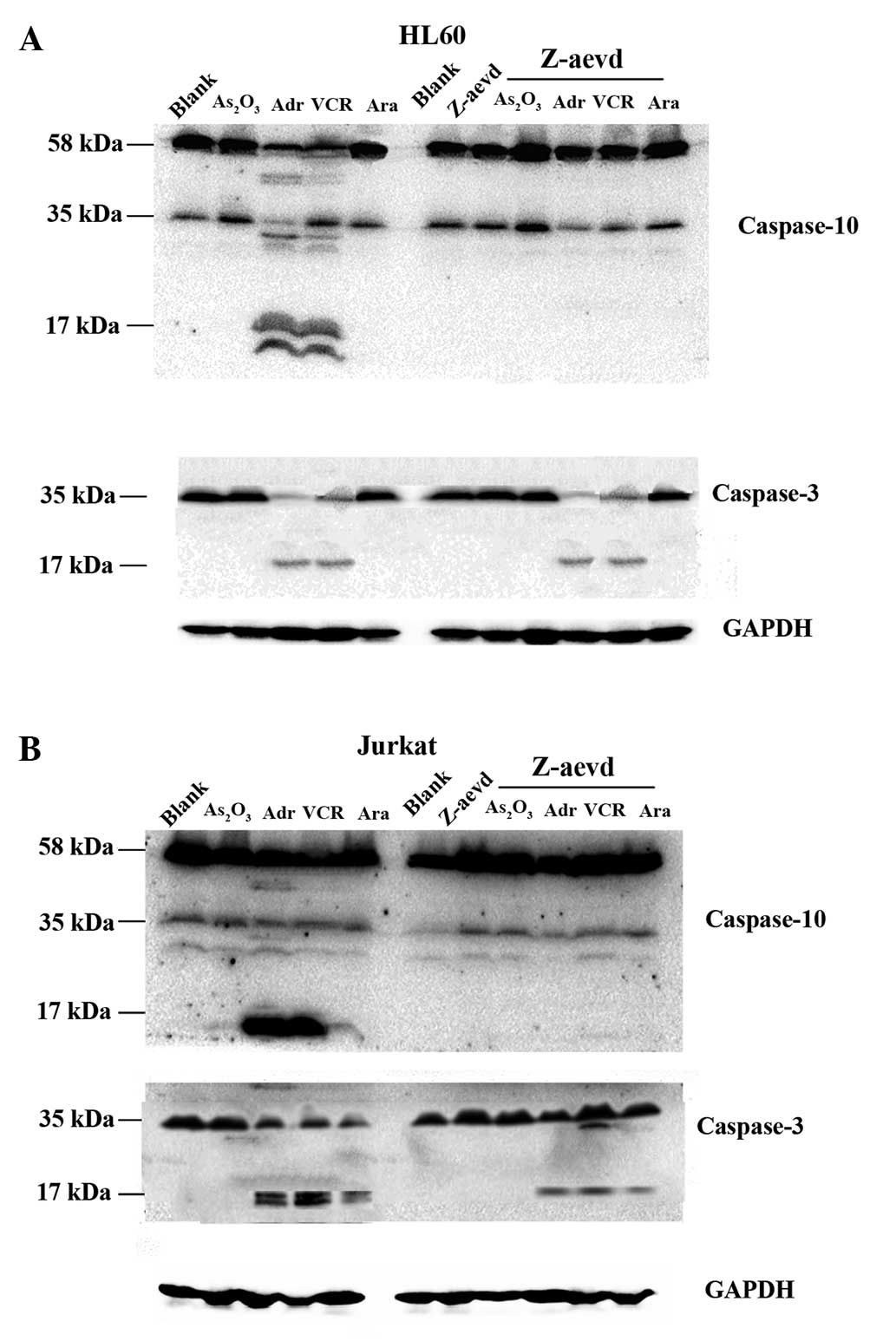
Levels of caspase-3 and −10 in HL-60
and Jurkat cells treated with either chemotherapy drugs or
chemotherapy drugs combined with a caspase-10 inhibitor
The expression of caspase-3 and −10 decreased in
HL-60 cells when the cells were treated with chemotherapy drugs
compared with the control group, and the cleaved forms of caspase-3
and −10 increased in the ADM and VCR groups. When the cells were
treated with chemotherapy drugs combined with the caspase-10
inhibitor, the expression of caspase-3 and −10 were similar to the
group treated with the chemotherapy drugs alone (Fig. 3A). This result also showed the change
in survival rate could not explained by apoptosis in HL-60
cells.
The expression of caspase-3 and −10 decreased in
Jurkat cells when the cells were treated with chemotherapy drugs
compared with the blank group, and the cleaved forms of caspase-3
and −10 increased in the ADM, VCR and Ara-c treated groups. When
the cells were treated with the chemotherapy drugs in combination
with the caspase-10 inhibitor, the expression of caspase-3 and −10
increased, and the cleaved forms of caspase-3 and −10 decreased
more compared with the group treated with chemotherapy drugs alone
(Fig. 3B). This result also showed
the change in survival rate could be explained by apoptosis in
Jurkat cells.
Survival rate of HL-60 and Jurkat
cells treated with chemotherapy drugs combined with a caspase-10
inhibitor and 3-MA
As the change in rate of apoptosis was not
significant in HL-60 cells treated in various ways, the present
study considered that the survival rate may be associated with ACD.
Thus, the autophagy inhibitor, 3-MA, was used. The survival rates
of HL-60 cells treated with chemotherapy drugs combined with the
caspase-10 inhibitor and 3-MA were: AS2O3,
74.82±1.02; ADM, 62.37±1.30; VCR, 54.09±2.03; and Ara-c, 20.2±3.2.
The results showed that the survival rate was increased compared
with the group treated with chemotherapy drugs combined with the
caspase-10 inhibitor. The survival rate also increased in HL-60
cells treated with the caspase-10 inhibitor combined with 3-MA,
compared with the group treated with caspase-10 inhibitor alone
(P=0.01; Table II; Fig. 1A). The results showed that the change
in survival rate was associated with ACD.
| Table II.Survival and apoptotic rates of HL-60
and Jurkat cells treated with chemotherapy drugs combined with
z-aevd-fmk and 3-MA for 24 h. |
Table II.
Survival and apoptotic rates of HL-60
and Jurkat cells treated with chemotherapy drugs combined with
z-aevd-fmk and 3-MA for 24 h.
|
| HL-60 cell | Jurkat cell |
|---|
|
|
|
|
|---|
| Treatment | Survival (%) | Apoptosis (%) | Survival (%) | Apoptosis (%) |
|---|
| Control |
102.55±9.46a |
0.51±0.11b |
100.62±8.73a |
0.62±0.12a |
| Z+control |
68.87±4.31a |
0.50±0.30b |
83.77±3.87a |
0.53±0.10b |
| 3-MA+Z+blank |
89.12±3.25a |
1.21±0.20b |
50.61±4.78 |
5.03±0.30a |
|
3-MA+Z+AS2O3 |
74.82±1.02a |
4.83±2.10b |
50.39±4.80 |
5.62±1.10a |
| 3-MA+Z+ADM |
62.37±1.30a |
9.00±2.30b |
44.99±3.67 |
22.20±5.80a |
| 3-MA+Z+VCR |
54.09±2.03a |
10.01±3.20b |
39.38±4.01 |
30.20±2.30a |
| 3-MA+Z+Ara-c |
77.17±5.65a |
2.61±0.90b |
54.32±2.34 |
7.20±1.10a |
|
Z+AS2O3 |
67.83±4.81a |
3.61±1.80b |
79.27±1.83a |
1.10±0.90a |
| Z+ADM |
33.89±2.48a |
9.01±3.40b |
54.01±2.85a |
12.21±5.60a |
| Z+VCR |
42.70±3.70a |
10.50±5.40b |
44.86±1.05a |
14.30±5.40a |
| Z+Ara-c |
69.11±1.11a |
3.62±1.10b |
57.23±0.49a |
6.72±2.10a |
The same test was also performed on the Jurkat cells
group. The survival rates of the Jurkat cells treated with
chemotherapy drugs combined with the caspase-10 inhibitor and 3-MA
were: AS2O3, 50.39±4.80; ADM, 44.99±3.67;
VCR, 39.38±4.01; and Ara-c, 54.32±2.34. The results showed that the
survival rate decreased compared with the group treated with
chemotherapy drugs combined with the caspase-10 inhibitor. The
survival rate also decreased in the Jurkat cells treated with the
caspase-10 inhibitor combined with 3-MA compared with the group
treated with the caspase-10 inhibitor alone (P=0.013; Table II; Fig.
1B). The result showed that treatment with 3-MA could result in
the decreased survival rate of Jurkat cells, which was different
from the result found in HL-60 cells.
Rate of apoptosis in HL-60 and Jurkat
cells treated with chemotherapy drugs combined with a caspase-10
inhibitor and 3-MA
The rates of apoptosis in HL-60 cells treated with
chemotherapy drugs combined with the caspase-10 inhibitor and 3-MA
for 24 h were: AS2O3, 4.8±2.1; ADM, 9.0±2.3;
VCR, 10.0±3.2; and Ara-c, 2.6±0.9. There was no significant
difference between the group treated with chemotherapy drugs
combined with the caspase-10 inhibitor and 3-MA and the group
treated with the chemotherapy drugs combined with the caspase-10
inhibitor. The rate of apoptosis in the group treated with 3-MA or
caspase-10 inhibitor alone was extremely low (P=0.095; Table II; Fig.
2A), and there was no significant difference between the group
treated with 3-MA and the group treated with the caspase-10
inhibitor (P=0.21; Table II;
Fig. 2A).
The rates of apoptosis in Jurkat cells treated with
chemotherapy drugs combined with the caspase-10 inhibitor and 3-MA
for 24 h were: AS2O3, 5.6±1.1; ADM, 22.2±5.8;
VCR, 30.2±2.3; and Ara-c, 6.7±1.1. The rate of apoptosis increased
significantly in the group treated with chemotherapy drugs combined
with caspase-10 inhibitor and 3-MA compared with the group treated
with the chemotherapy drugs combined with the caspase-10 inhibitor
(P=0.21; Table II; Fig. 2B). The rate of apoptosis in the group
treated only with 3-MA was increased compared with the rate of
apoptosis in the group treated with only the caspase-10 inhibitor
(P=0.011; Table II; Fig. 2B).
Discussion
Autophagy is critical for maintaining normal
cellular homeostasis; basal levels of autophagy are required to
maintain cellular fitness and the quality control of essential
cellular components, by removing unfolded, excessive or aged
proteins, as well as damaged or superfluous organelles (2,19–21). Cancer may be affected by defects in
apoptosis, but the association between autophagy and cancer is
complex. Whether autophagy is a promoter or killer of cancer cells
remains a subject of discussion (22). Certain studies have indicated that
autophagy could protect cancer cells when they are treated with
chemotherapy (23,24), and others suggest that autophagy may
be associated with chemotherapy or radiation therapy resistance
(25,26). However, other studies have mentioned
that the excessive activation of autophagy could result in the ACD
of cancer cells (27), or contribute
to ACD through unchecked degradative processes (7–9). From
reviewing the literature, the authors of the present study have
concluded autophagy may be a promoter or killer of cancer cells
depending on the context. Thus, discovering the factors that
regulate autophagy is key to manipulating autophagy for cancer
therapy.
Lamy et al found an association between
capase10 and autophagy in myeloma cells (14). Therefore, it may be hypothesized that
caspase-10 is also important in autophagy in AL cells. In the
present study, the survival rate of HL-60 cells decreased and the
survival rate of Jurkat cells increased after the cells were
treated with chemotherapy drugs combined with a caspase-10
inhibitor. However, the survival rate of the HL-60 and Jurkat cells
decreased after the cells were treated with only the caspase-10
inhibitor. Therefore, tests were performed on the rate of apoptosis
and the expression level of caspase-3 to attempt to explain the
change in the survival rate. These tests showed differences between
the HL-60 and the Jurkat cells. The rate of apoptosis in the HL-60
cells did not change after the cells were treated with chemotherapy
drugs combined with the caspase-10 inhibitor compared with the
group treated with chemotherapy drugs alone. Caspase-3 analysis
also showed that apoptosis was not activated after the cells were
treated with chemotherapy drugs combined with the caspase-10
inhibitor. The results also indicated that the caspase-10 inhibitor
did not activate apoptosis by itself. So the decreasing survival
rate could not be explained by apoptosis in HL-60 cells. However,
the change in the rate of apoptosis and the expression levels of
caspase-3 in Jurkat cells showed that apoptosis was inhibited after
the cells were treated with chemotherapy drugs combined with the
caspase-10 inhibitor. According to these results, the increasing
survival rate in Jurkat cells could be potentially caused by the
inhibition apoptosis. However, the decreased Jurkat cell survival
rate following treatment with only the caspase-10 inhibitor remains
unexplained, since the caspase-10 inhibitor did not activate
apoptosis by itself in Jurkat cells.
These results indicated that another type of cell
death may be occurring in the HL-60 cells and in the Jurkat cells
that were treated with only the caspase-10 inhibitor. Autophagy has
been classified as a form of programmed cell death, termed ACD.
This term describes a form of caspase-independent necrosis-like
cell death associated with the accumulation of autophagosomes
(27). Thus, the change in the
survival rate may potentially be caused by ACD. 3-MA, which can
inhibit autophagy, was added to the set of HL-60 cells treated with
the chemotherapy drugs combined with the caspase-10 inhibitor, or
treated with only the caspase-10 inhibitor. The survival rate
increased when 3-MA was added compared with the group treated with
chemotherapy drugs combined with the caspase-10 inhibitor, or
treated with the caspase-10 inhibitor only, and the rate of
apoptosis did not decrease. The results also indicated that the
rate of apoptosis was extremely low when the HL-60 cells were
treated with 3-MA alone. Thus, caspase-10 could be associated with
the basal level of autophagy in HL-60 cells, and ACD may increase
in HL-60 cells when caspase-10 is inhibited. However, the survival
rate remained decreased in the Jurkat cells when treated with the
caspase-10 inhibitor in combination with 3-MA, as an increased rate
of apoptosis was indicated compared with the rate of apoptosis in
HL-60 cells. Therefore, the change in the survival rate in the
Jurkat cells that had been treated with the caspase-10 inhibitor
only cannot be explained by autophagy, and further investigation is
required to explain this phenomenon. When 3-MA was added to the
Jurkat cell group treated with chemotherapy drugs, the decrease in
survival rate was associated with an increasing rate of apoptosis.
This result is similar to the results of the study on Jurkat cells
by Liu et al (16), which
indicated that inhibiting autophagy could improve the chemotherapy
effect on leukemia cells. According to these results, the different
role of caspase-10 in HL-60 and Jurkat cells may be associated with
the various biological characteristics of HL-60 and Jurkat cells.
Lamy et al (14) showed that
caspase-10 does not play the same role in lymphoma cells as it does
in myeloma cells, and that the biological characteristics of Jurkat
cells are more similar to lymphatic cells. This may be the reason
for the different results in HL-60 and Jurkat cells.
As the present study has only preliminarily shown
that caspase-10 could be associated with autophagy, the next stage
of research would be to investigate the association between
caspase-10 and autophagy in acute myeloid leukemia cells, and to
determine the mechanisms or signaling channels for the
caspase-10-associated regulation of autophagy in acute myeloid
leukemia.
In conclusion, the role of caspase-10 may be
associated with the basal levels of autophagy in acute myeloid
leukemia cells, particularly when the cells meet stressful
conditions, such as chemotherapy treatment. When caspase-10 is
inhibited, ACD appeared in acute myeloid leukemia cells. However,
there was no evidence to support a role for caspase-10 in autophagy
in the acute lymphoid leukemia cells used in the present study.
These results suggest a novel role for caspase-10 in the regulation
of acute myeloid leukemia cell death and a novel method for the
treatment of acute myeloid leukemia.
Acknowledgements
The authors would like to thank Mrs. Chen Hui and
Mr. Yang Junjun for their skillful technical assistance, and
Professor Zhu Xueqiong for funding assistance (all The Second
Affiliated Hospital and Yuying Children's Hospital, Wenzhou,
Zhejiang, China). The present study was supported by a program from
the Wenzhou Science and Technology Bureau, Wenzhou, China (grant
no. Y20140276).
References
|
1
|
Klionsky DJ, Cuervo AM, Dunn WA Jr, Levine
B, van der Klei I and Seglen PO: How shall I eat thee? Autophagy.
3:413–416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ma T, Zhu J, Chen X, Zha D, Singhal PC and
Ding G: High glucose induces autophagy in podocytes. Exp Cell Res.
319:779–789. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Evangelisti C, Chiarini F, Lonetti A,
Buontempo F, Neri LM, McCubrey JA, McCubrey JA and Martelli AM:
Autophagy in acute leukemias: A double-edged sword with important
therapeutic implications. Biochim Biophys Acta. 1853:14–26. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagelkerke A, Bussink J, Geurts-Moespot A,
Sweep FC and Span PN: Therapeutic targeting of autophagy in cancer.
Part II: Pharmacological modulation of treatment-induced autophagy.
Semin Cancer Biol. 31:99–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Macintosh RL and Ryan KM: Autophagy in
tumour cell death. Semin Cancer Biol. 23:344–351. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pullarkat V, Meng Z, Donohue C, Yamamoto
VN, Tomassetti S, Bhatia R, Krishnan A, Forman SJ and Synold TW:
Iron chelators induce autophagic cell death in multiple myeloma
cells. Leuk Res. 388:988–996. 2014. View Article : Google Scholar
|
|
8
|
Ristic B, Bosnjak M, Arsikin K, Mircic A,
Suzin-Zivkovic V, Bogdanovic A, Perovic V, Martinovic T,
Kravic-Stevovic T, Bumbasirevic V, et al: Idarubicin induces
mTOR-dependent cytotoxic autophagy in leukemic cells. Exp Cell Res.
326:90–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He W, Wang Q, Srinivasan B, Xu J, Padilla
MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, et al: A JNK-mediated
autophagy pathway that triggers c-IAP degradation and necroptosis
for anticancer chemotherapy. Oncogene. 33:3004–3013. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Galluzzi L, Kepp O, Krautwald S, Kroemer G
and Linkermann A: Molecular mechanisms of regulated necrosis. Semin
Cell Dev Biol. 35:24–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lorin S, Hamai A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang SF, Wang XL, Yang XQ and Chen N:
Autophagy-associated targeting pathways of natural products during
cancer treatment. Asian Pac J Cancer Prev. 15:10557–10563. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lamy L, Ngo VN, Emre NC, Shaffer AL III,
Yang Y, Tian E, Nair V, Kruhlak MJ, Zingone A, Landgren O and
Staudt LM: Control of autophagic cell death by caspase-10 in
multiple myeloma. Cancer Cell. 23:435–449. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wilson NS, Dixit V and Ashkenazi A: Death
receptor signal transducers: Nodes of coordination in immune
signaling networks. Nat Immunol. 10:348–355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang SP, Niu YN, Yuan N, Zhang AH, Chao
D, Xu QP, Wang LJ, Zhang XG, Zhao WL, Zhao Y and Wang JR: Role of
autophagy in acute myeloid leukemia therapy. Chin J Cancer.
32:130–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Watson AS, Mortensen M and Simon AK:
Autophagy in the pathogenesis of myelodysplastic syndrome and acute
myeloid leukemia. Cell Cycle. 10:1719–1725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuma A, Hatano M, Matsui M, Yamamoto A,
Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T and Mizushima N: The
role of autophagy during the early neonatal starvation period.
Nature. 432:1032–1036. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boya P, González-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Morselli E, Galluzzi L, Kepp O, Vicencio
JM, Criollo A, Maiuri MC and Kroemer G: Anti- and pro-tumor
functions of autophagy. Biochim Biophys Acta. 1793:1524–1532. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Degenhardt K, Mathew R, Beandoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato K, Tsuehihara K, Fujii S, Sugiyama M,
Goya T, Atomi Y, Ueno T, Ochiai A and Esumi H: Autophagy is
activated in colorectal cancer cells and contributes to the
tolerance to nutrient deprivation. Cancer Res. 67:9677–9684. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pel AA, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shimizu S, Kanaseki T, Mizushima N, Mizuta
T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y: Role of Bcl-2
family proteins in a non-apoptopic programmed cell death dependent
on autophagy genes. Nat Cell Biol. 6:1221–1228. 2004. View Article : Google Scholar : PubMed/NCBI
|