Introduction
Renal cell carcinoma (RCC) originates from
epithelial cells of the proximal tubule and is the most common type
of kidney cancer worldwide (1). The
incidence rates among males are 6.6 in the UK, 9.7 in Germany, and
10 in the USA (per 100,000); incidence rates vary substantially
across different countries or regions, for example 2.8 in Korea and
15.3 in the Czech Republic. Rates among females are approximately
half of those among males (1). Due to
its high resistance to conventional radiation and chemotherapy,
metastatic RCC has a very poor prognosis. Significant efforts have
been made in the last two decades to identify the genetic basis of
RCC. In the majority of cases of sporadic RCC, deletion or mutation
of the tumor suppressor gene Von-Hippel-Lindau (VHL) has been
detected (2). VHL catalyzes the
degradation of hypoxia-inducible factor-α (HIF-α) under normoxic
conditions, and loss of VHL results in accumulation of HIF-α and
downstream induction of vascular endothelial growth factor (VEGF),
as well as other factors that contribute to tumorigenesis (3). Targeting VEGF signaling pathways has
increased the therapeutic options in the treatment of metastatic
RCC (4). However, continued research
is necessary to gain a better insight into the biological basis of
carcinogenesis and metastasis, and to identify potential targets
for novel therapeutic strategies.
Cancer cells produce a variety of chemokines
(5). Chemokine secretion recruits
inflammatory and immune cells to the tumor site; in the majority of
cases, this process is associated with tumor progression and
metastasis, as infiltrating inflammatory and immune cells
facilitate tumor cell proliferation, angiogenesis, repression of
the adaptive immune response and degradation of the extracellular
matrix (6). One of the most important
chemokines in this process is monocyte chemoattractant protein-1
[MCP-1; also known as chemokine (C-C motif) ligand 2], a member of
the C-C chemokine superfamily, which regulates leukocyte
recruitment primarily via the chemokine (C-C motif) receptor type 2
(CCR2) (7). In addition to the
recruitment of inflammatory cells, MCP-1 also induces angiogenesis
by chemoattraction of CCR2-expressing endothelial cells (8,9). High
expression levels of MCP-1 in tumor cells and leukocyte recruitment
has been described in various cancer types, including mammary
(10), ovarian (11), pancreatic (12) and prostate (13) cancer.
Several studies have also addressed the role of
MCP-1/CCR2 signaling in RCC. In patients exhibiting RCC, enhanced
MCP-1 expression in tumor cells, and infiltration of the tumors
with tumor-associated macrophages and tumor-infiltrating
lymphocytes has been observed (14,15). In
xenograft models of RCC, enhanced MCP-1 expression is associated
with microvessel density and tumor size (16,17).
In prostate carcinoma cells, in addition to
paracrine signaling to recruit inflammatory or endothelial cells,
autocrine MCP-1 signaling to promote cell proliferation and
invasiveness has also been observed (18). Deletion of VHL stimulates MCP-1
expression, possibly via VEGF signaling (16,17,19);
however, the mechanism of regulation of MCP-1 expression in
VHL+/+ RCC is largely unknown. Therefore, the
present study examined the regulation and function of MCP-1 in the
VHL+/+ RCC cell line, CaKi-1, and the
VHL−/− RCC cell line, 786-O. Particular
attention was paid to a possible role of osmosensitive
transcription factor nuclear factor of activated T cells 5 (NFAT5;
also known as tonicity enhancer binding protein or osmotic response
element binding protein), which stimulates MCP-1 expression in
kidney epithelial cells during inflammatory processes (20,21). Our
previous study reported that cellular NFAT5 activity is enhanced in
CaKi-1 cells, a phenomenon associated with increased expression of
the NFAT5 target gene, S100 calcium binding protein A4 (S100A4;
also known as metastasin), resulting in an enhanced proliferation
and migration ability of these cells (22).
The aim of the present study was to determine
whether autocrine MCP-1 signaling has a role in RCC cells and
whether there are differences in the regulation of MCP-1 expression
between different RCC cell lines. The present study provides
evidence that autocrine MCP1/CCR2 signaling stimulates
proliferation and migration of RCC cells, and that NFAT5 mediates
the expression of MCP-1 in the VHL+/+ cell
line CaKi-1, but not in the VHL−/− cell line
786-O.
Materials and methods
Materials
Rabbit polyclonal anti-NFAT5 antibody was purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA; dilution,
1:1,000; catalog no., sc-13035); rabbit polyclonal anti-β-actin
antibody was purchased from Sigma-Aldrich (Deisenhofen, Germany;
dilution 1:5,000; catalog no., A2066); goat anti-rabbit polyclonal
horseradish peroxidase-conjugated anti-rabbit IgG antibody was
purchased from Cell Signaling Technology (Beverly, MA, USA;
dilution, 1:5,000; catalog no., 7074). Human MCP-1 enzyme-linked
immunosorbent assay (ELISA) kit (catalog no., 900-K31), MCP-1
neutralizing antibody and recombinant human (rh) MCP-1 were
obtained from PeproTech (Hamburg, Germany). PVP-free polycarbonate
track etched filters were obtained from GVS S.p.A. (Bologna,
Italy). Accell SMARTpool siRNA for knockdown of NFAT5 and Accell
non-targeting siRNA #2 were obtained from Thermo Fisher
Scientific, Inc. (Epsom, UK). The CCR2 inhibitor, RS504393, and
ethidium bromide were obtained from Sigma-Aldrich. DNA ladder ‘Mass
Ruler’ was purchased from Thermo Fisher Scientific, Inc. Agarose
was obtained from Bioline (Luckenwalde, Germany) and cell culture
plates were obtained from Greiner-Bio One GmbH (Frickenhausen,
Germany).
Cell culture
HK-2 (CRL-2190) immortalized human proximal tubule
cells, and CaKi-1 (HTB-46) and 786-O (CRL-1932) RCC cells, all
purchased from American Type Culture Collection (Manassas, VA,
USA), were cultured in Dulbecco's modified Eagle medium (DMEM;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (Biochrom GmbH, Berlin, Germany), 100 U/ml penicillin and 100
µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.,
Karlsruhe, Germany). Cells were grown at 37°C in a humidified
atmosphere (95% air; 5% CO2). Human peripheral blood
mononuclear cells (PBMCs) were freshly prepared from the
heparinized blood samples of healthy donors using Leucosep tubes
(Greiner Bio-One GmbH), according to the manufacturer's
instructions. Blood samples were obtained in accordance with German
federal law.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
For determination of MCP-1, CCR2, NFAT5 and β-actin
mRNA expression levels, total RNA was isolated from CaKi-1, 786-O
and HK-2 cells using TriFast reagent (Peqlab Biotechnologie GmbH,
Erlangen, Germany), according to the manufacturer's instructions.
The concentration of RNA in each sample was determined
spectrophotometrically at 260 nm. Absence of RNA degradation was
tested by denaturing agarose gel electrophoresis on a 1.5%
agarose/formaldehyde gel, checking for clear sharp bands for 28S
and 18S rRNA, and absence of excessive smear. To avoid
amplification of genomic DNA, intron-spanning primer pairs were
designed for RT-qPCR analysis. The primers (Metabion International
AG, Martinsried, Germany) used were as follows: Forward,
5′-AGTCTCTGCCGCCCTTCT-3′ and reverse, 5′-GTGACTGGGGCATTGATTG-3′ for
MCP-1; forward, 5′-CTGTCCACATCTCGTTCTCGGTTTA-3′ and reverse,
5′-CCCAAAGACCCACTCATTTGCAGC-3′ for CCR2; forward,
5′-AATCGCCCAAGTCCCTCTAC-3′ and reverse, 5′-GGTGGTAAAGGAGCTGCAAG-3′
for NFAT5; and forward, 5′-CCAACCGCGAGAAGATGA-3′ and reverse,
5′-CCAGAGGCGTACAGGGATAG-3′ for β-actin. The efficiency of each
primer pair was tested in initial experiments. Standard curves were
generated using standards of 100, 10, 1 and 0.1 ng total starting
RNA. In experiments comparing relative mRNA levels by RT-qPCR, the
RNA concentration of the samples was adjusted to 25 ng/µl and
constant amounts of RNA (75 ng) were used in each experiment. To
ensure that the expression of the reference gene (β-actin) was
stable among all three cell lines and all experimental conditions,
Cq values of β-actin were compared and found no significant
differences. Experiments were conducted on a CFX Connect Real Time
PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA)
using the SensiMix SYBR One-Step kit (Bioline; catalog no., BIO
98005), according to the manufacturers' instructions. The relative
mRNA expression levels of each gene was calculated using the
2−ΔΔCq method (23), with
β-actin as the housekeeping gene. Cq values were corrected for PCR
efficiency according to the following formula: CqE= Cq *
[log(E) / Log(2)], where E is
efficiency and 100% efficiency is 2. Negative controls lacking
reverse transcriptase enzyme or RNA template were used in the
experiments to exclude that traces of genomic DNA or other DNA
contaminations were amplified. Specificity of PCR product formation
was confirmed by monitoring melting point analysis and by agarose
gel electrophoresis. Each experiment was repeated six times.
MCP-1 concentration
CaKi-1, 786-O and HK-2 cells were grown in 96-well
plates, as described. After reaching confluency, growth medium was
replaced by serum-free DMEM and cells were incubated for a further
48 h. The concentration of MCP-1 in the cell culture supernatant
was determined using the specific ELISA kit, according to the
manufacturers' instructions. To normalize for differences in cell
number, cells from each well were collected after removal of the
culture supernatant, the total DNA content/well was determined
spectrophotometrically at an absorbance of 260 nm, and MCP-1
concentration was normalized to DNA content.
Proliferation assays
For proliferation assays, CaKi-1 and 786-O cells
were seeded in 96-well plates at a density of 5×103
cells/well and incubated for 24 h for cell attachment, as
described. Thereafter, growth medium was removed and cells were
grown for another 24–96 h in serum-free DMEM in the presence of rh
MCP-1 (10 ng/ml), MCP-1 neutralizing antibody (1 µg/ml) or CCR2
antagonist RS504393 (10 µM). Control cells were left untreated.
Relative cell numbers in each well were determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (24). Briefly, cells were
incubated with MTT (final concentration, 0.5 mg/ml in serum-free
DMEM) for 4 h at 37°C. Thereafter, the medium was removed, formazan
crystals were solubilized in 100 µl acidified isopropanol and
absorption was measured at 565 nm on an Infinite M200 Pro
microplate reader (Tecan Deutschland GmbH, Crailsheim,
Germany).
Migration assays
Migration of PBMCs towards the conditioned medium
(CM) of RCC cells was analyzed in a modified Boyden chamber
(25). To obtain CM, CaKi-1 and 786-O
cells were grown to 100% confluency in 24-well plates, as
described. Subsequently, the growth medium was replaced by
serum-free DMEM and cells were incubated for a further 48 h. The
cell culture CM was collected by pipetting and centrifuged at
12,000 × g for 5 mins to remove unattached cells or cell debris;
subsequently, the supernatant was collected. To adjust for
differences in cell number, cells from each well were collected
after removal of CM and total DNA content/well was determined
spectrophotometrically (absorbance at 260 nm). CM were then
normalized for DNA content between samples by adding serum-free
DMEM. The CM was added to the lower compartments of the Boyden
chamber for experimental samples, while controls were filled with
unconditioned serum-free DMEM. In some experiments, the CM was
preincubated with an MCP-1 neutralizing antibody (1 µg/ml) for 1 h
at room temperature. The lower compartments were separated from the
upper compartments by a porous polycarbonate membrane (pore size, 5
µm). The upper compartments were each filled with 105
freshly prepared PBMCs dissolved in serum-free DMEM. In some
experiments, PBMCs were preincubated with CCR2 antagonist RS504393
(10 µM) for 1 h at room temperature. The Boyden chamber was
incubated for 4 h at 37°C and PBMCs that had migrated across the
polycarbonate membrane into the lower compartment were counted
using a hemocytometer under an inverted light microscope.
Cell migration of CaKi-1 and 786-O cells was
analyzed using the in vitro scratch assay (26), also known as the wound healing assay.
CaKi-1 and 786-O cells were grown to confluency, as described.
Subsequently, the growth medium was removed and the cell monolayers
were ‘scratched’ with a 200-µl pipette tip. Cells were further
incubated in serum-free DMEM in the presence of MCP-1 neutralizing
antibody (1 µg/ml), CCR2 antagonist RS504393 (10 µM) or rh MCP-1
(10 ng/ml); control cells were left untreated. Cell migration into
the immediate vicinity of the scratch was monitored using an
inverted microscope (IM35; Zeiss, Oberkochen, Germany) and by
capturing images every 4 h.
Knockdown of NFAT5
CaKi-1 or 786-O cells were grown to ~80% confluency,
trypsinized, washed in PBS and finally resuspended in 100 µl
modified HEPES-buffered saline electroporation buffer (0.5% HEPES,
1% glucose, 0.5% Ficoll, 5 mM NaCl, 135 mM KCl, 2 mM
MgCl2, pH 7.4) containing 2 µM Accell SMARTpool NFAT5
siRNA or Accell non-targeting siRNA #2 (control).
Electroporation was conducted with a Gene Pulser Xcell
Electroporation System (Bio-Rad Laboratories) at 140 V and 1,000 µF
(exponential decay pulse) in a 2-mm cuvette and the cells seeded
immediately thereafter in 24-well plates. Cells were incubated for
5 days prior to western blot analysis. Knockdown efficiency was
determined by densitometric analysis of western blots using ImageJ
version 1.47 software (National Institutes of Health, Bethesda, MD,
USA)
Immunoblot analysis
The cells were seeded in 24-well plates and washed
three times with chilled PBS. The cells were collected by scraping
and lysed by the addition of 50 µl urea (8 M)/PBS, followed by
three freeze/thaw cycles and finally centrifuged at 12,000 × g for
5 min at 4°C. Supernatants were used as whole-cell-protein lysates.
Protein concentration in the lysates was measured by the Bradford
method (27), using a commercially
available assay (Bio-Rad Laboratories), according to the
manufacturer's protocol. Aliquots (5–30 µg protein) were subjected
to 10% SDS-PAGE (28) and blotted
onto nitrocellulose membranes (GE Healthcare, Pittsburgh, PA, USA).
Non-specific binding sites were blocked with 5% non-fat dry milk in
phosphate-buffered saline (PBS) containing 0.1% Tween-20 (PBS-T) at
room temperature for 1 h. Samples were incubated with primary
antibodies in PBS-T containing 5% non-fat dry milk overnight at
4°C. Subsequently, the blots were washed 3 times with PBS-T for 5
min each, and the membranes incubated with the appropriate
secondary antibody at room temperature for 1 h in PBS-T containing
5% non-fat dry milk. After washing with PBS-T 3 times for 5 min
each, immunocomplexes were visualized by enhanced chemiluminescence
(Pierce ECL Western Blotting Substrate; Thermo Fisher Scientific,
Inc.).
Statistical analysis
Statistical analysis was performed using SPSS
version 18.0 software (SPSS, Inc., Chicago, IL, USA). Data are
expressed as means ± standard error of the mean. The differences
between the means were assessed by two-way analysis of variance
followed by Tukey's post hoc test. P<0.05 was used to indicate a
statistically significant difference. All experiments were
performed at least 3 times and representative results are
shown.
Results
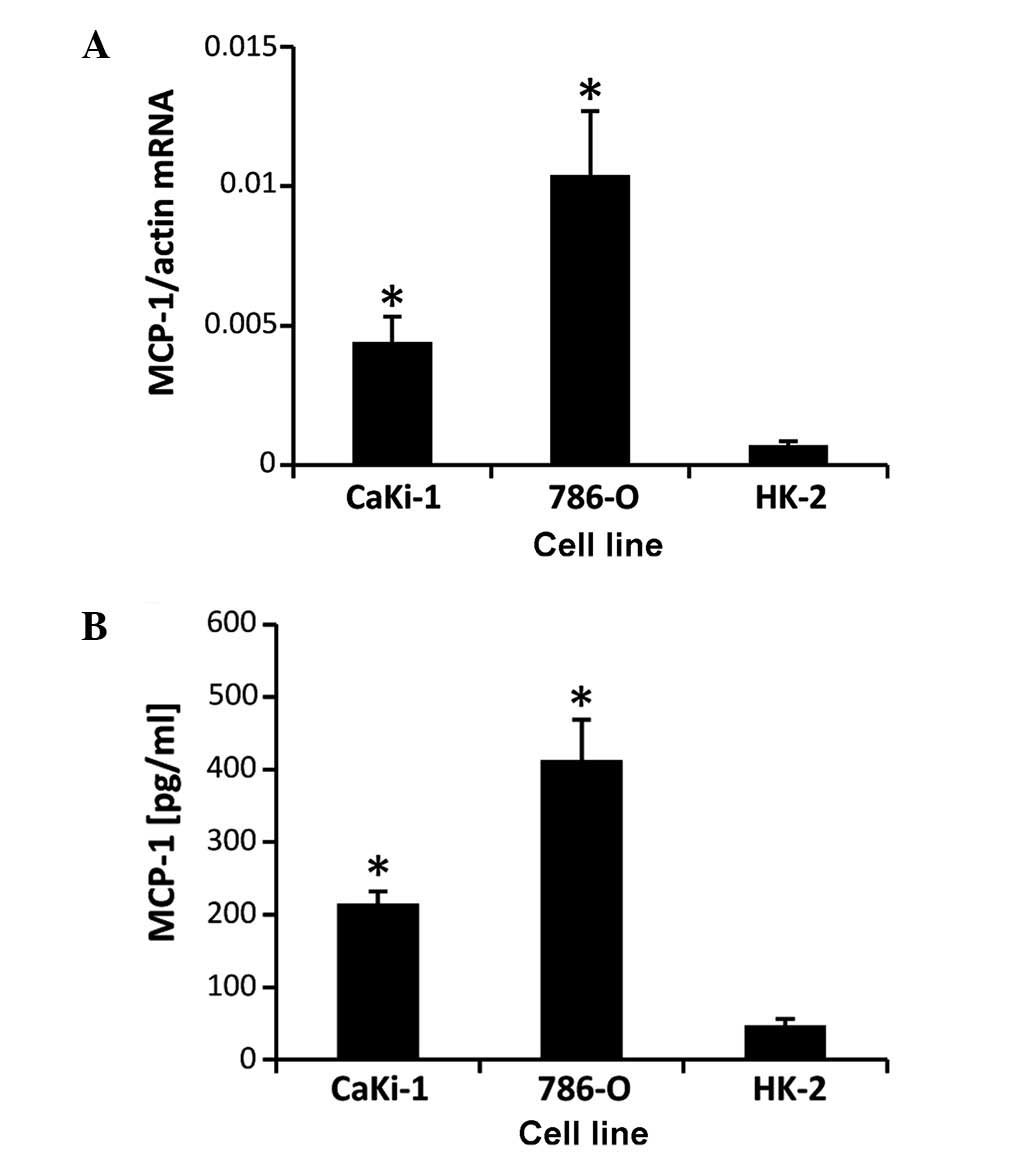
MCP-1 is highly expressed in RCC
cells
CaKi-1 and 786-O cell lines were used as models for
metastatic clear cell RCC. The proximal tubule cell line, HK-2, was
used as non-cancerous control cells. Expression of MCP-1 was
determined by RT-qPCR (Fig. 1A) and
ELISA assay (Fig. 1B). The results
showed that MCP-1 mRNA and protein is expressed in all three cell
lines, however, CaKi-1 and 786-O cells exhibited ~5-fold and
~12-fold increases in expression levels, respectively, compared
with HK-2 cells, demonstrating that MCP-1 expression is enhanced in
RCC cells.
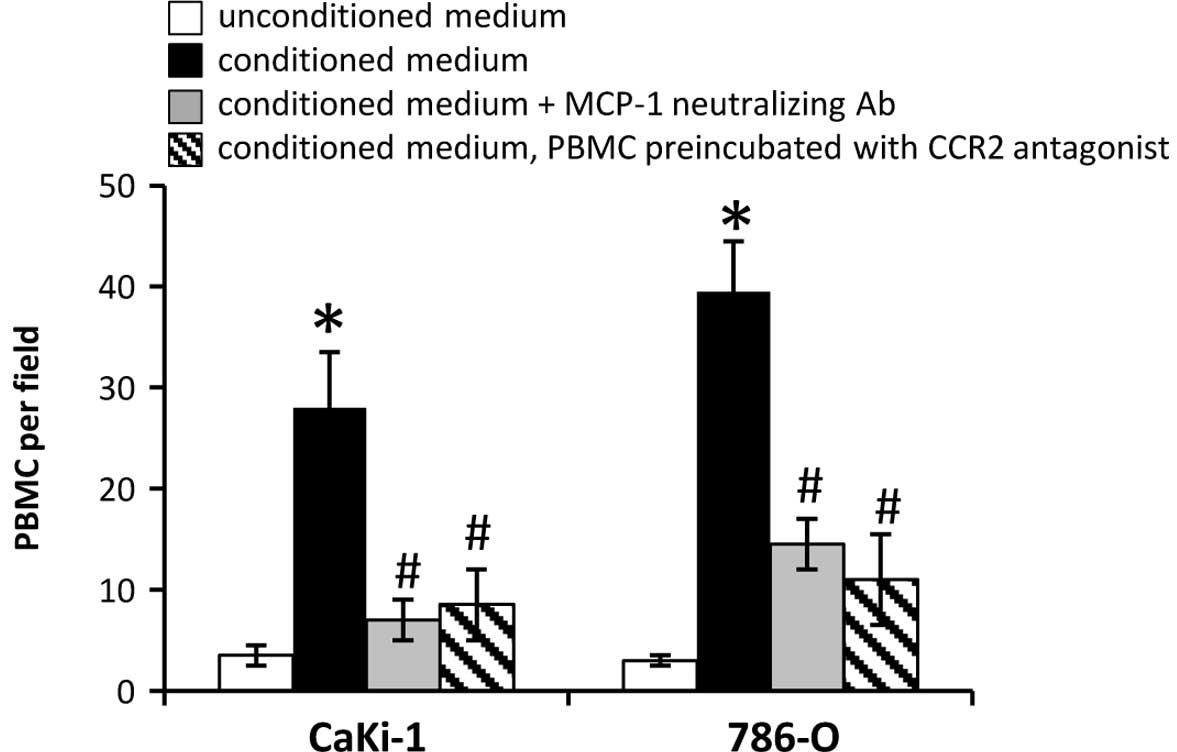
MCP-1 secretion of RCC cells mediates
monocyte recruitment
Secretion of MCP-1 by tumor cells and subsequent
recruitment of circulating blood monocytes has an important role
during infiltration of the tumor stroma by inflammatory cells in
various cancer types. The recruitment of PBMCs to conditioned,
serum-free CM of CaKi-1 or 786-O cells was examined using a
modified Boyden chamber. As expected, monocyte recruitment towards
the CM of both cell lines was significantly stronger than towards
unconditioned medium (P<0.05; Fig.
2). The results also demonstrate that the migration of
monocytes towards the CM of 786-O cells, which express higher
concentrations of MCP-1 than CaKi-1 cells (Fig. 1), was stronger than towards the CM of
CaKi-1 cells (P<0.05; Fig. 2).
Furthermore, preincubation of the CM of both cell lines with an
MCP-1 neutralizing antibody and preincubation of PBMCs with a CCR2
antagonist (RS504393) both significantly diminished monocyte
recruitment compared with CM alone (P<0.05; Fig. 2). These data indicate that MCP-1
secreted by RCC cells attracts PBMCs via binding to CCR2.
Autocrine effects of MCP-1 on the
proliferation and migration of RCC cells
In prostate carcinoma cells, autocrine binding of
MCP-1 to CCR2 stimulates their proliferation and migration ability
(18). Expression of CCR2 in CaKi-1
and 786-O cells was detected by RT-qPCR in both cell lines. No
significant differences in CCR2 expression levels were observed
between the two cell lines (P>0.05; Fig. 3A). The presence of CCR2 suggests that
autocrine MCP-1/CCR2 signaling may also occur in RCC cells. To test
this hypothesis, cells were grown in serum-free medium and treated
with an MCP-1 neutralizing antibody, rh MCP-1 or a CCR2 antagonist
(RS504393). Control cells were left untreated. Cell numbers were
determined at 24-h intervals between 24 and 96 h by MTT assay. As
shown in Fig. 3B, incubation with a
CCR2 antagonist or neutralizing antibody significantly decreased
the proliferation of 786-O and CaKi-1 cells compared with untreated
control cells. However, additional supplementation of the growth
medium with rh MCP-1 had no significant effect on cell growth
compared with the control cells.
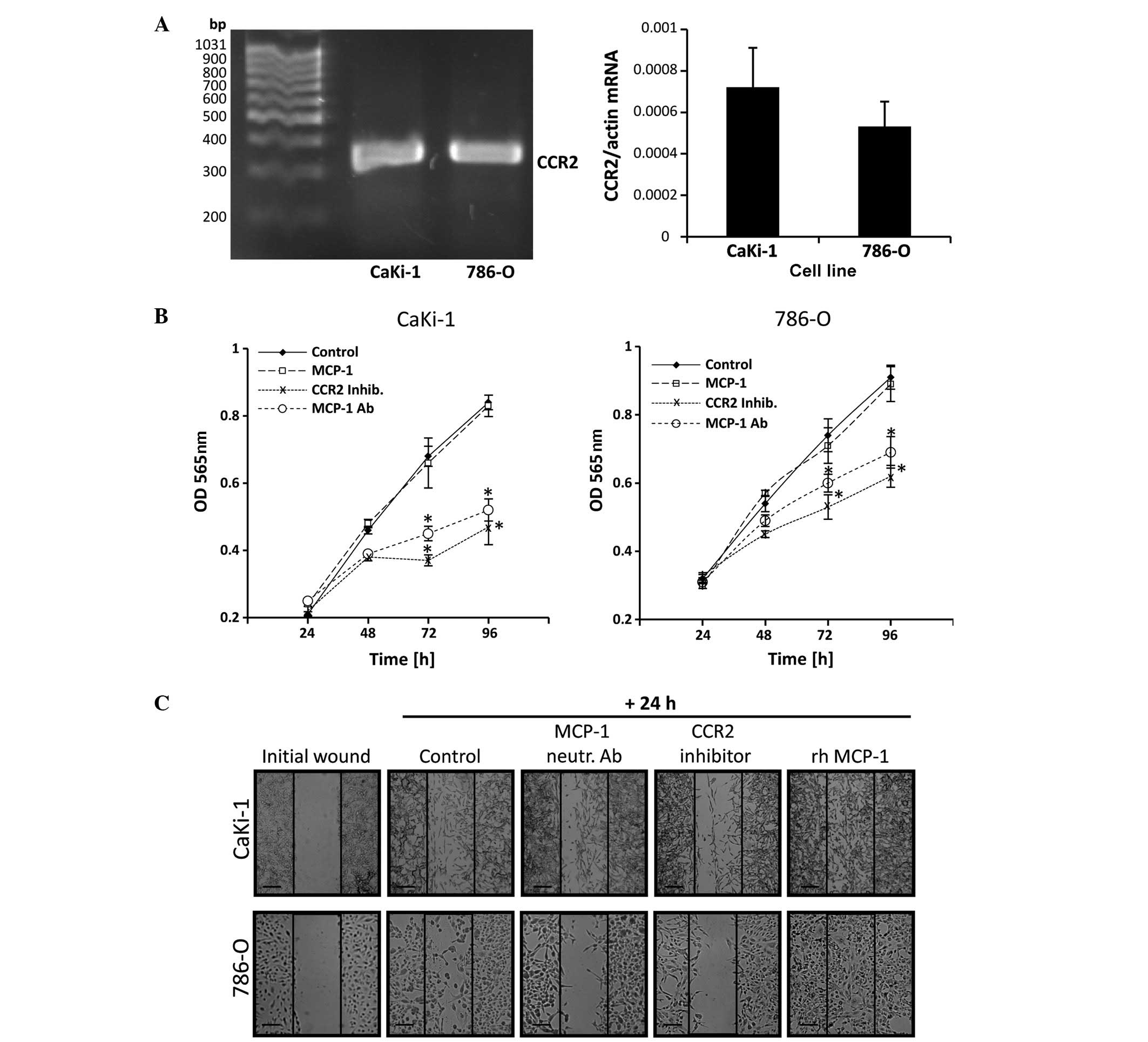 | Figure 3.Autocrine MCP-1/CCR2 signaling
stimulates the proliferation and migration of renal cell carcinoma
cells. (A) RNA from 786-O and CaKi-1 cells was extracted and CCR2
expression was determined by reverse transcription-quantitative
polymerase chain reaction. The correct size of the amplification
product (324 bp) was verified by agarose gel electrophoresis.
Relative CCR2 mRNA abundance was normalized to that of β-actin to
correct for differences in RNA input. Data are presented as the
mean ± standard error of the mean (SEM; n=6). (B) To determine
proliferation, CaKi-1 or 786-O cells (5×103/well) were
seeded into a 96-well plate and incubated for 24 h for cell
attachment. Thereafter, cells were grown for another 24–96 h in
serum-free Dulbecco's modified Eagle medium and relative cell
numbers in each well determined by MTT assay. Cells were grown in
the presence of recombinant human (rh) MCP-1 (□), an MCP-1
neutralizing antibody (○) or the CCR2 antagonist RS504393 (x). The
control cells were left untreated (♦). Data are presented as the
mean ± SEM (n=8). *P<0.05 vs. untreated control. (C) To
determine migration, confluent CaKi-1 or 786-O cells were scratched
with a 200-µl pipette tip and then incubated for another 24 h.
Cells were incubated in the presence of rh MCP-1, an MCP-1
neutralizing antibody or the CCR2 inhibitor RS504393. Control cells
were left untreated. Shown are representative phase-contrast images
of cells migrating into the wounded area, immediately after
scratching and after 24 h incubation. Images were obtained at ×40
magnification on a Zeiss IM35 inverted microscope. Scale bars, 100
µm. CCR2, chemokine (C-C motif) receptor type 2; OD, optical
density; MCP-1, monocyte chemoattractant protein-1; inhib,
inhibitor; Ab, antibody; rh, recombinant human. |
The migration ability of RCC cells was determined by
performing a scratch assay. A monolayer of confluent CaKi-1 or
786-O cells was scratched with a pipette tip and cells were treated
with MCP-1 neutralizing antibody, rh MCP-1 or CCR2 antagonist
(RS504393). Control cells were left untreated. Cell migration into
the immediate vicinity of the scratch was observed under a
microscope. As indicated in Fig. 3C,
migration of RCC cells into the wounded area in the presence of
CCR2 antagonist or neutralizing MCP-1 antibody was markedly
decreased compared with control cells after 24 h. By contrast,
supplementation with recombinant MCP-1 enhanced cell migration
ability.
These results indicate that autocrine MCP-1/CCR2
signaling stimulates the proliferation and migration of RCC cells.
Endogenous MCP-1 expression levels appear to be sufficient for the
maximum proliferation rate but not for the maximum migratory
ability of RCC cells.
NFAT5 knockdown decreases MCP-1
expression in CaKi-1, but not in 786-O cells
MCP-1 expression in renal tubular cells is known to
be regulated by the osmosensitive transcription factor NFAT5
(20,21), which is highly expressed in CaKi-1
cells (22). To test the hypothesis
that the high expression levels of MCP-1 in CaKi-1 cells are a
result of high NFAT5 activity in RCC cells, NFAT5 mRNA and protein
expression, and the effect of siRNA-mediated NFAT5 knockdown on
MCP-1 expression were examined in CaKi-1 and 786-O cells. As shown
in Fig. 4A and B, NFAT5 mRNA and
protein expression, respectively, were significantly lower in 786-O
cells than CaKi-1 cells (P<0.05), with their expression
comparable to that in HK-2 cells. Transfection of CaKi-1 and 786-O
cells with an NFAT5-specific siRNA construct resulted in a
reduction of NFAT5 protein expression by 86 and 79%, respectively,
compared with cells transfected with non-specific control siRNA
(P<0.05; Fig. 4C). Furthermore,
NFAT5 knockdown significantly decreased the expression of MCP-1 in
CaKi-1 cells (P<0.05), but had no significant impact on MCP-1
expression levels in 786-O cells (Fig. 4D
and E). These results indicate that MCP-1 expression depends
largely on NFAT5 in CaKi-1 cells, but not in 786-O cells.
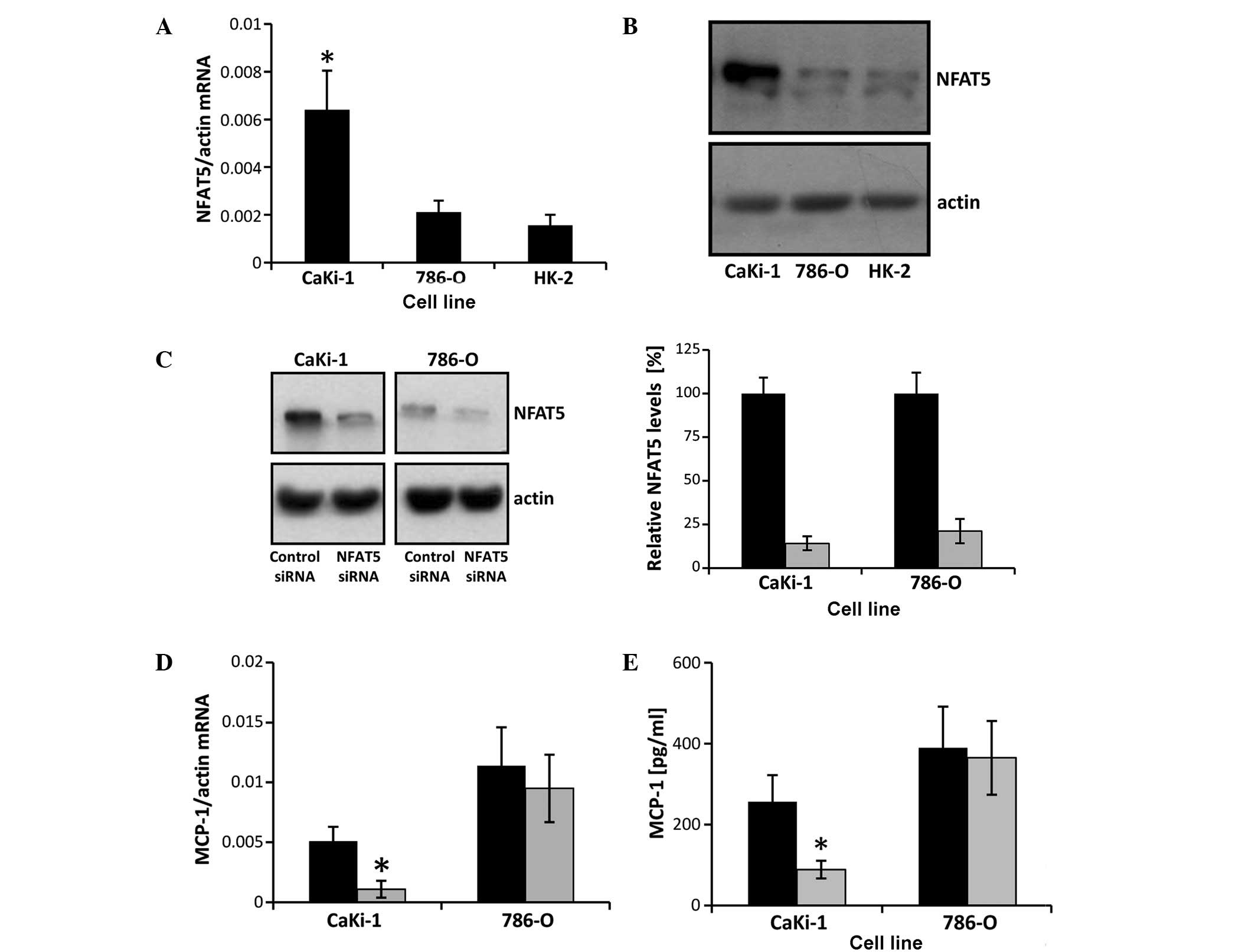 | Figure 4.NFAT5 knockdown attenuates MCP-1
expression in CaKi-1 but not in 786-O cells. (A) RNA from CaKi-1,
786-O and HK-2 cells was extracted, and the abundance of NFAT5 and
β-actin mRNA transcripts determined by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
Relative mRNA abundance of NFAT5 was normalized to that of β-actin
to correct for differences in RNA input. Data are presented as mean
± standard error of the mean (SEM; n=6). P<0.05 vs.
HK-2 control cells. (B) Protein lysates from CaKi-1, 786-O and HK-2
cells were processed for immunoblotting to determine the expression
of NFAT5. To demonstrate comparable protein loading, the blots were
also probed for β-actin. Representative blots from 4 independent
experiments are shown. (C) CaKi-1 and 786-O cells were transfected
with siRNA constructs specific for NFAT5 (■) or with non-targeting
siRNA (■) as controls, and were grown for 72 h. Thereafter, protein
lysates were processed for immunoblotting to confirm NFAT5
knockdown. To demonstrate comparable protein loading, the blots
were also probed for β-actin. Representative blots from 4
independent experiments are shown. Densitometric analysis of NFAT5
abundance was performed using ImageJ software. NFAT5 expression in
CaKi-1 or 786-O cells transfected with non-targeting siRNA was
defined as 100%. Data are presented as the mean ± SEM (n=4). (D)
CaKi-1 and 786-O cells were transfected with siRNA constructs
specific for NFAT5 (■) or with non-targeting siRNA (■) as controls,
and were grown for 72 h. To determine MCP-1 transcription levels,
RNA was extracted from the cells and the abundance of MCP-1 mRNA
was determined by RT-qPCR. Relative MCP-1 mRNA abundance was
normalized to that of β-actin to correct for differences in RNA
input. Data are presented as the mean ± SEM (n=4). *P<0.05 vs.
CaKi-1 cells transfected with non-targeting control siRNA. (E)
CaKi-1 and 786-O cells were transfected with siRNA constructs
specific for NFAT5 (■) or with non-targeting siRNA (■) as controls,
and were grown for 72 h. To determine MCP-1 secretion, cells were
subsequently incubated for 48 h in serum-free Dulbecco's modified
Eagle medium. Thereafter, samples were collected and the
concentration of MCP-1 in the cell culture supernatant was
determined by enzyme-linked immunosorbent assay. Data are presented
as the mean ± SEM (n=6). *P<0.05 vs. CaKi-1 transfected with
non-targeting control siRNA. NFAT5, nuclear factor of activated T
cells 5; MCP-1, monocyte chemoattractant protein-1. |
Discussion
High expression levels of the chemokine MCP-1 have
been observed in various types of tumor cell and MCP-1/CCR2
signaling is known to have an important role in tumor progression
(29). To date, relatively few
studies have addressed this signaling cascade in RCC. Yamasaki
et al (17) used xenograft
models to demonstrate that the expression of MCP-1 in RCC cells is
correlated with tumor growth. The protumorigenic effects of MCP-1
are typically explained by the angiogenic activity and enhanced
recruitment of inflammatory cells, which in turn secrete a variety
of growth factors into the tumor microenvironment. The present
study provides evidence that, in addition to this paracrine MCP-1
signaling, autocrine MCP-1 signaling also occurs in RCC cells, and
may contribute to tumor progression by promoting cell proliferation
and cell migration. In the current study, the expression of MCP-1
and its receptor, CCR2, were detected in 786-O and CaKi-1 RCC
cells. Neutralization of MCP-1 by a specific antibody or
pharmacological inhibition of CCR2 significantly decreased cell
growth in both cell lines. The results of the current study agree
with a previous study, in which prostate carcinoma cell
proliferation was stimulated by autocrine MCP-1/CCR2 signaling
(18,30). Therefore, another important mechanism
for the development of metastatic RCC may be the positive effect of
autocrine MCP-1/CCR2 signaling on the cell migration ability of
CaKi-1 and 786-O cells.
The results of the present study also indicate that
endogenous MCP-1 expression levels are sufficient for maximum
proliferation, but not for maximum migration of RCC cells. These
limitations regarding migration ability may be overcome during
metastasis by an increase in MCP-1 expression (31). Metastatic RCC is characterized by
frequent bone metastases (32).
During metastasis, osteoblast-derived factors increase MCP-1
expression in RCC cells to enhance cancer cell migration via an
autocrine mechanism, thereby facilitating bone metastasis (31). This process is mediated via the
cancer-associated cell membrane glycoprotein, dysadherin. Data from
the literature suggest that a similar mechanism occurs during bone
metastasis of mammary carcinoma cells (33), indicating that autocrine MCP-1/CCR2
signaling may also have an important role during bone metastasis in
other types of cancer.
The importance of autocrine MCP-1/CCR2 signaling,
particularly during metastasis of RCC cells, is also highlighted by
the fact that only 15% of primary tumor cells but 52% of metastatic
cells express CCR2 (34). These data
suggest that CCR2 may be a potential target for future treatment
strategies of metastatic RCC (and other cancer types), as
pharmacological inhibition of CCR2 may not only influence paracrine
MCP-1 signaling to decrease the recruitment of inflammatory
monocytes and endothelial cells, but also affect autocrine MCP-1
signaling, thereby decreasing the proliferation and migration
ability of carcinoma cells. The first clinical trials investigating
the therapeutic potential of pharmacological CCR2 inhibitors in
pancreatic cancer are currently in progress (35).
For future studies, it will be important to evaluate
the proliferative and pro-migratory pathways that are activated by
autocrine MCP-1/CCR2 signaling in cancer cells. It is well
documented that CCR2 activates the phosphatidyl inositol 3 kinase
(PI3K)/Akt pathway (30), which in
turn can stimulate cell proliferation, for example via activation
of the mammalian target of rapamycin signaling pathway (36). CCR2 can also stimulate cell migration,
for example via activation of p70S6 kinase (37). However, due to the complexity of the
PI3K/Akt signaling pathway, extensive analysis of downstream
signaling molecules will be necessary to reveal the detailed
molecular mechanisms of autocrine MCP-1/CCR2 signaling in cancer
cells.
The results of the present and previous studies
indicate that there are substantial differences in the regulation
of MCP-1 expression between VHL−/− RCC cell
lines, such as 786-O, and VHL+/+ cells, such
as CaKi-1 (16,17,19), and
this may have an impact on the progression and treatment of RCC.
Our previous study showed that, in CaKi-1 cells, NFAT5 activity is
increased (22) and MCP-1 expression
largely depends on this increased NFAT5 activity, as supported by
the results of the present study. By contrast, NFAT5 is not
increased in 786-O cells compared with non-cancerous HK-2 cells,
and MCP-1 expression is appears to be mediated by VEGF signaling in
786-O cells and other VHL−/− RCC cell lines
(19). The expression of MCP-1 is
generally higher in VHL−/− cells than in
VHL+/+ cells (17). Accordingly, enhanced MCP-1 expression
is associated with increased angiogenesis and tumor growth in 786-O
murine xenografts compared with CaKi-1 xenografts. Treatment of
this 786-O murine xenograft with the VEGF-targeting monoclonal
antibody, bevacizumab, significantly decreased angiogenesis and
tumor growth, while CaKi-1 xenografts were virtually unaffected by
this therapy (17). Therefore, drugs
that target the VEGF signaling pathway and are already approved for
the treatment of metastatic RCC, such as bevacizumab, sunitinib or
sorafenib, may elicit their anti-tumorigenic effects in part by
decreasing MCP-1 expression in VHL−/− RCC,
but may not affect auto- and paracrine MCP-1/CCR2 signaling in
VHL+/+ RCC.
NFAT5 is another potential target for future
treatment strategies of VHL+/+ RCC and other
tumor entities. Under physiological conditions, NFAT5 regulates the
expression of several osmoprotective and urinary concentrating
genes in the renal medulla (38–42), as
well as the expression of pro-inflammatory cytokines in macrophages
and lymphocytes (43,44). Several studies suggest that NFAT5 has
an important role in the development of various cancer types, such
as non-small cell lung cancer (45,46),
melanoma (47), leiomyoma (48), breast cancer (49–51) and
colon carcinoma (52–54). A recent study identified NFAT5 as a
master regulator of inflammatory breast cancer (55), which is the most aggressive type of
breast cancer due to its high angiogenic potential, invasiveness,
and frequent local and metastatic recurrences. An important target
gene of NFAT5 in the context of tumor progression is S100A4, as
demonstrated in colon, breast and RCC cells (22,51,52,56).
The present study provides evidence that, in addition to S100A4,
MCP-1 may also be upregulated by NFAT5 in carcinoma cells. The
upregulation of MCP-1 by dysadherin has an important role during
bone metastasis of cancer cells; however, the molecular mechanisms
by which dysadherin enhances MCP-1 expression are largely unknown.
Notably, dysadherin is a modulator of Na+/K+-ATPase (57,58), which
has been shown to stimulate NFAT5 activity (59–61). As
NFAT5 and nuclear factor-κB (NF-κB), which is also activated by
dysadherin (33), can cooperatively
induce MCP-1 expression (21,62), we propose that dysadherin stimulates
MCP-1 expression via co-activation of NFAT5 and NF-κB. However, the
details of this mechanism remain to be elucidated.
In conclusion, the present study demonstrated that
autocrine MCP-1/CCR2 signaling stimulates the proliferation and
migration ability of RCC cells. MCP-1 expression in the
VHL+/+ cell line, CaKi-1, but not in the
VHL−/− cell line, 786-O, is mediated by the
transcription factor NFAT5, further emphasizing the role of NFAT5
as a protumorigenic factor. The present results suggest that
MCP-1/CCR2 axis and/or NFAT5 may act as potential targets for
future therapeutic strategies in the treatment of metastatic
RCC.
Acknowledgements
The present study was supported by grants from the
Deutsche Forschungsgemeinschaft (grant no., NE839/6-1), the
Münchener Medizinische Wochenschrift and the Friedrich Baur
Foundation (grant no., FBS05/11) (Munich, Germany). The authors
thank Dr John Davis and Dr Christopher Batters for critical reading
of the manuscript. The technical assistance of Mrs. Maria-Luisa
Fraek is gratefully acknowledged.
References
|
1
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Linehan WM, Walther MM and Zbar B: The
genetic basis of cancer of the kidney. J Urol. 170:2163–2172. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patel PH, Chadalavada RS, Chaganti RS and
Motzer RJ: Targeting von Hippel-Lindau pathway in renal cell
carcinoma. Clin Cancer Res. 12:7215–7220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Motzer RJ: New perspectives on the
treatment of metastatic renal cell carcinoma: An introduction and
historical overview. Oncologist. 16(Suppl 2): S1–S3. 2011.
View Article : Google Scholar
|
|
5
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Solinas G, Germano G, Mantovani A and
Allavena P: Tumor-associated macrophages (TAM) as major players of
the cancer-related inflammation. J Leukoc Biol. 86:1065–1073. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deshmane SL, Kremlev S, Amini S and Sawaya
BE: Monocyte chemoattractant protein-1 (MCP-1): An overview. J
Interferon Cytokine Res. 29:313–326. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Salcedo R, Ponce ML, Young HA, Wasserman
K, Ward JM, Kleinman HK, Oppenheim JJ and Murphy WJ: Human
endothelial cells express CCR2 and respond to MCP-1: Direct role of
MCP-1 in angiogenesis and tumor progression. Blood. 96:34–40.
2000.PubMed/NCBI
|
|
9
|
Weber KS, Nelson PJ, Gröne HJ and Weber C:
Expression of CCR2 by endothelial cells: Implications for MCP-1
mediated wound injury repair and in vivo inflammatory activation of
endothelium. Arterioscler Thromb Vasc Biol. 19:2085–2093. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ueno T, Toi M, Saji H, Muta M, Bando H,
Kuroi K, Koike M, Inadera H and Matsushima K: Significance of
macrophage chemoattractant protein-1 in macrophage recruitment,
angiogenesis, and survival in human breast cancer. Clin Cancer Res.
6:3282–3289. 2000.PubMed/NCBI
|
|
11
|
Negus RP, Stamp GW, Hadley J and Balkwill
FR: Quantitative assessment of the leukocyte infiltrate in ovarian
cancer and its relationship to the expression of C-C chemokines. Am
J Pathol. 150:1723–1734. 1997.PubMed/NCBI
|
|
12
|
Sanford DE, Belt BA, Panni RZ, Mayer A,
Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA
and Goetz BD: Inflammatory monocyte mobilization decreases patient
survival in pancreatic cancer: A role for targeting the CCL2/CCR2
axis. Clin Cancer Res. 19:3404–3415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Loberg RD, Ying C, Craig M, Yan L, Snyder
LA and Pienta KJ: CCL2 as an important mediator of prostate cancer
growth in vivo through the regulation of macrophage infiltration.
Neoplasia. 9:556–562. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Daurkin I, Eruslanov E, Stoffs T, Perrin
GQ, Algood C, Gilbert SM, Rosser CJ, Su LM, Vieweg J and Kusmartsev
S: Tumor-associated macrophages mediate immunosuppression in the
renal cancer microenvironment by activating the 15-lipoxygenase-2
pathway. Cancer Res. 71:6400–6409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ferrero E, Fabbri M, Poggi A, Galati G,
Bernasconi S and Zocchi MR: Tumor-driven matrix invasion by
infiltrating lymphocytes: Involvement of the alpha1 integrin
I-domain. Eur J Immunol. 28:2530–2536. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kanno T, Kamba T, Yamasaki T, Shibasaki N,
Saito R, Terada N, Toda Y, Mikami Y, Inoue T, Kanematsu A, et al:
JunB promotes cell invasion and angiogenesis in VHL-defective renal
cell carcinoma. Oncogene. 31:3098–3110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamasaki T, Kamba T, Kanno T, Inoue T,
Shibasaki N, Arakaki R, Yamada T, Kondo K, Kamoto T, Nishiyama H,
et al: Tumor microvasculature with endothelial fenestrations in VHL
null clear cell renal cell carcinomas as a potent target of
anti-angiogenic therapy. Cancer Sci. 103:2027–2037. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu Y, Cai Z, Galson DL, Xiao G, Liu Y,
George DE, Melhem MF, Yao Z and Zhang J: Monocyte chemotactic
protein-1 (MCP-1) acts as a paracrine and autocrine factor for
prostate cancer growth and invasion. Prostate. 66:1311–1318. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li C, Liu B, Dai Z and Tao Y: Knockdown of
VEGF receptor-1 (VEGFR-1) impairs macrophage infiltration,
angiogenesis and growth of clear cell renal cell carcinoma (CRCC).
Cancer Biol Ther. 12:872–880. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kojima R, Taniguchi H, Tsuzuki A, Nakamura
K, Sakakura Y and Ito M: Hypertonicity-induced expression of
monocyte chemoattractant protein-1 through a novel cis-acting
element and MAPK signaling pathways. J Immunol. 184:5253–5262.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roth I, Leroy V, Kwon HM, Martin PY,
Féraille E and Hasler U: Osmoprotective transcription factor
NFAT5/TonEBP modulates nuclear factor-kappaB activity. Mol Biol
Cell. 21:3459–3474. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Küper C, Beck FX and Neuhofer W:
NFAT5-mediated expression of S100A4 contributes to proliferation
and migration of renal carcinoma cells. Front Physiol. 5:2932014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Falk W, Goodwin RH Jr and Leonard EJ: A
48-well micro chemotaxis assembly for rapid and accurate
measurement of leukocyte migration. J Immunol Methods. 33:239–247.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Conti I and Rollins BJ: CCL2 (monocyte
chemoattractant protein-1) and cancer. Semin Cancer Biol.
14:149–154. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Loberg RD, Day LL, Harwood J, Ying C, St
John LN, Giles R, Neeley CK and Pienta KJ: CCL2 is a potent
regulator of prostate cancer cell migration and proliferation.
Neoplasia. 8:578–586. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schuler Y, Lee-Thedieck C, Geiger K,
Kaiser T, Ino Y, Aicher WK and Klein G: Osteoblast-secreted factors
enhance the expression of dysadherin and CCL2-dependent migration
of renal carcinoma cells. Int J Cancer. 130:288–299. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Coleman RE: Clinical features of
metastatic bone disease and risk of skeletal morbidity. Clin Cancer
Res. 12(Suppl): S6243–S6249. 2006. View Article : Google Scholar
|
|
33
|
Nam JS, Kang MJ, Suchar AM, Shimamura T,
Kohn EA, Michalowska AM, Jordan VC, Hirohashi S and Wakefield LM:
Chemokine (C-C motif) ligand 2 mediates the prometastatic effect of
dysadherin in human breast cancer cells. Cancer Res. 66:7176–7184.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wyler L, Napoli CU, Ingold B, Sulser T,
Heikenwälder M, Schraml P and Moch H: Brain metastasis in renal
cancer patients: Metastatic pattern, tumour-associated macrophages
and chemokine/chemoreceptor expression. Br J Cancer. 110:686–694.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang-Gillam A, Nywening TM, Sanford DE,
Lockhart AC, Suresh R, Tan BR, Lim KH, Sorscher S, Fowler K, Amin
MA, et al: Phase IB study of FOLFIRINOX plus PF-04136309 in
patients with borderline resectable and locally advanced pancreatic
adenocarcinoma (PC). J Clin Oncol. 33(Suppl; Abstract
388)2015.PubMed/NCBI
|
|
36
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qian Y, Corum L, Meng Q, Blenis J, Zheng
JZ, Shi X, Flynn DC and Jiang BH: PI3K induced actin filament
remodeling through Akt and p70S6K1: Implication of essential role
in cell migration. Am J Physiol Cell Physiol. 286:C153–C163. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miyakawa H, Woo SK, Chen CP, Dahl SC,
Handler JS and Kwon HM: Cis- and trans-acting factors regulating
transcription of the BGT1 gene in response to hypertonicity. Am J
Physiol. 274:F753–F761. 1998.PubMed/NCBI
|
|
39
|
Miyakawa H, Woo SK, Dahl SC, Handler JS
and Kwon HM: Tonicity-responsive enhancer binding protein, a
rel-like protein that stimulates transcription in response to
hypertonicity. Proc Natl Acad Sci USA. 96:2538–2542. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Z, Ferraris JD, Brooks HL, Brisc I
and Burg MB: Expression of osmotic stress-related genes in tissues
of normal and hyposmotic rats. Am J Physiol Renal Physiol.
285:F688–F693. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Woo SK, Lee SD, Na KY, Park WK and Kwon
HM: TonEBP/NFAT5 stimulates transcription of HSP70 in response to
hypertonicity. Mol Cell Biol. 22:5753–5760. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Han KH, Woo SK, Kim WY, Park SH, Cha JH,
Kim J and Kwon HM: Maturation of TonEBP expression in developing
rat kidney. Am J Physiol Renal Physiol. 287:F878–F885. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
López-Rodriguez C, Aramburu J, Jin L,
Rakeman AS, Michino M and Rao A: Bridging the NFAT and NF-kappaB
families: NFAT5 dimerization regulates cytokine gene transcription
in response to osmotic stress. Immunity. 15:47–58. 2001.PubMed/NCBI
|
|
44
|
Esensten JH, Tsytsykova AV,
Lopez-Rodriguez C, Ligeiro FA, Rao A and Goldfeld AE: NFAT5 binds
to the TNF promoter distinctly from NFATp, c, 3 and 4, and
activates TNF transcription during hypertonic stress alone. Nucleic
Acids Res. 33:3845–3854. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhong L, Peng X, Hidalgo GE, Doherty DE,
Stromberg AJ and Hirschowitz EA: Identification of circulating
antibodies to tumor-associated proteins for combined use as markers
of non-small cell lung cancer. Proteomics. 4:1216–1225. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mijatovic T, Mathieu V, Gaussin JF, De
Nève N, Ribaucour F, Van Quaquebeke E, Dumont P, Darro F and Kiss
R: Cardenolide-induced lysosomal membrane permeabilization
demonstrates therapeutic benefits in experimental human non-small
cell lung cancers. Neoplasia. 8:402–412. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Levy C, Khaled M, Iliopoulos D, Janas MM,
Schubert S, Pinner S, Chen PH, Li S, Fletcher AL, Yokoyama S, et
al: Intronic miR-211 assumes the tumor suppressive function of its
host gene in melanoma. Mol Cell. 40:841–849. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
McCarthy-Keith DM, Malik M, Britten J,
Segars J and Catherino WH: Gonadotropin-releasing hormone agonist
increases expression of osmotic response genes in leiomyoma cells.
Fertil Steril. 95:2383–2387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Germann S, Gratadou L, Zonta E, Dardenne
E, Gaudineau B, Fougère M, Samaan S, Dutertre M, Jauliac S and
Auboeuf D: Dual role of the ddx5/ddx17 RNA helicases in the control
of the pro-migratory NFAT5 transcription factor. Oncogene.
31:4536–4549. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jauliac S, López-Rodriguez C, Shaw LM,
Brown LF, Rao A and Toker A: The role of NFAT transcription factors
in integrin-mediated carcinoma invasion. Nat Cell Biol. 4:540–544.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen M, Sinha M, Luxon BA, Bresnick AR and
O'Connor KL: Integrin alpha6beta4 controls the expression of genes
associated with cell motility, invasion, and metastasis, including
S100A4/metastasin. J Biol Chem. 284:1484–1494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen M, Sastry SK and O'Connor KL: Src
kinase pathway is involved in NFAT5-mediated S100A4 induction by
hyperosmotic stress in colon cancer cells. Am J Physiol Cell
Physiol. 300:C1155–C1163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Alvarez-Diaz S, Valle N, Ferrer-Mayorga G,
Lombardía L, Herrera M, Domínguez O, Segura MF, Bonilla F, Hernando
E and Muñoz A: MicroRNA-22 is induced by vitamin D and contributes
to its antiproliferative, antimigratory and gene regulatory effects
in colon cancer cells. Hum Mol Genet. 21:2157–2165. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Slattery ML, Lundgreen A, Bondurant KL and
Wolff RK: Tumor necrosis factor-related genes and colon and rectal
cancer. Int J Mol Epidemiol Genet. 2:328–338. 2011.PubMed/NCBI
|
|
55
|
Remo A, Simeone I, Pancione M, Parcesepe
P, Finetti P, Cerulo L, Bensmail H, Birnbaum D, Van Laere SJ,
Colantuoni V, et al: Systems biology analysis reveals NFAT5 as a
novel biomarker and master regulator of inflammatory breast cancer.
J Transl Med. 13:1382015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li JT, Wang LF, Zhao YL, Yang T, Li W,
Zhao J, Yu F, Wang L, Meng YL, Liu NN, et al: Nuclear factor of
activated T cells 5 maintained by Hotair suppression of miR-568
upregulates S100 calcium binding protein A4 to promote breast
cancer metastasis. Breast Cancer Res. 16:4542014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lubarski I, Asher C and Garty H: FXYD5
(dysadherin) regulates the paracellular permeability in cultured
kidney collecting duct cells. Am J Physiol Renal Physiol.
301:F1270–F1280. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lubarski I, Pihakaski-Maunsbach K, Karlish
SJ, Maunsbach AB and Garty H: Interaction with the Na,K-ATPase and
tissue distribution of FXYD5 (related to ion channel). J Biol Chem.
280:37717–37724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lim SW, Ahn KO, Sheen MR, Jeon US, Kim J,
Yang CW and Kwon HM: Downregulation of renal sodium transporters
and tonicity-responsive enhancer binding protein by long-term
treatment with cyclosporin A. J Am Soc Nephrol. 18:421–429. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jeon US, Han KH, Park SH, Lee SD, Sheen
MR, Jung JY, Kim WY, Sands JM, Kim J and Kwon HM: Downregulation of
renal TonEBP in hypokalemic rats. Am J Physiol Renal Physiol.
293:F408–F415. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Neuhofer W, Woo SK, Na KY, Grunbein R,
Park WK, Nahm O, Beck FX and Kwon HM: Regulation of TonEBP
transcriptional activator in MDCK cells following changes in
ambient tonicity. Am J Physiol Cell Physiol. 283:C1604–C1611. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Küper C, Beck FX and Neuhofer W: NFAT5
contributes to osmolality-induced MCP-1 expression in mesothelial
cells. Mediators Inflamm. 2012:5130152012. View Article : Google Scholar : PubMed/NCBI
|