Introduction
Serous papillary peritoneal carcinoma (SPPC) was
first described in 1959 by Swerdlow (1), and this was similar to ovarian papillary
cystadenocarcinoma with regard to the pathological features and to
pelvic peritoneal mesothelioma when considering diffused peritoneal
lesions. In 1993, extraovarian peritoneal serous papillary
carcinoma (EPSPC) was formally recognized according to
Gynecological Oncology Group (GOG) criteria in order to better
define the SPPC patient population (2). The clinical presentations of EPSPC
generally include abdominal distension and pain, ascites and
increasing levels of serum cancer antigen 125 (CA125), with the
absence of a pelvic mass on imaging examinations (2,3). Surgical
investigation and histopathological characteristics provide the
evaluation for a diagnosis of EPSPC in the light of GOG criteria:
Each ovary must be normally sized or enlarged by a benign process
(4.0 cm in largest diameter), and extraovarian site involvement
must be greater than that on the ovarian surface. Furthermore, the
ovarian component must be microscopically non-existent or confined
to the surface epithelium of the ovary, with no evidence of
cortical invasion, or must involve the ovarian epithelium and/or
the underlying stroma at <5×5 mm in depth and extent. The tumor
thus resembles serous papillary ovarian cancer (SPOC) in terms of
pathological features (2,4).
It is widely accepted that EPSPC is derived from the
mullerian coelomic epithelium, similar to serous ovarian carcinoma
(SOC) (5–7). However, histopathological and molecular
evidence is accumulating to show that peritoneal carcinomas are
multifocal in origin, thus differing from SOCs, which are only
unifocal. Considering the histological, molecular and clinical
similarities, EPSPC has traditionally been incorporated in the same
staging systems of stage III/IV SPOC and managed with the similar
pattern of surgical debulking and platinum-based chemotherapy as
used in advanced ovarian cancer (5,6,8).
EPSPC is indistinguishable from advanced ovarian
cancer in terms of clinical presentation, management and molecular
biology using immunohistochemistry or polymerase chain reaction
(PCR)-based assays (4). However,
preliminary genetic data from retrospective series have provided
certain indications that may be worthy of note and research; for
example, patterns of loss of heterozygosity (LOH) at a number of
chromosomal loci differ from those that are apparent in ovarian
cancer. Despite effective chemotherapeutic cytoreduction and
occasional long-term remissions (9),
patients with EPSPC survive for a shorter length of time than
ovarian cancer patients. Thus, any distinct molecular
characteristics in EPSPC are worth investigating, and are of great
importance for the illustration of unique disease characteristics.
Given the lack of comprehensive molecular data, the present study
performed whole-exome sequencing and comparative genomic
hybridization (CGH) analysis to investigate the molecular
characteristics of a case of EPSPC, which showed distinct genetic
alterations in the form of gene amplification and loss, somatic
mutations and germline mutations.
Materials and methods
Case identification
A 54-year-old woman was admitted to Yangpu Hospital
(Shanghai, China) in December 2012 due to a persistent low-grade
fever, shortness of breath and abdominal pain. Pelvic palpation
found that the patient's bilateral utero-sacral ligaments were
thickened with no existence of an actual mass. The serum CA125
level was increased significantly to 2,119 U/ml (normal level,
0.00–35.00 U/ml). In addition, the serum levels of CA153 (normal
range, 0.00–31.30 U/ml) and CA724 (normal range, 0.00–9.00 U/ml)
were slightly increased (43.50 and 23.47 U/ml, respectively),
whereas the levels of CA199 (normal range, 0.00–35.00 U/ml), CA242
(normal range, 0.05–20.00 U/ml), CA50 (normal range, 0.21–25.00
U/ml), squamous cell carcinoma antigen (normal range, 0.01–2.50
ng/ml), carcinoembryonic antigen (CEA; normal concentration,
<2.50 ng/ml) and α-fetoprotein (AFP; normal range, 0.00–10.00
ng/ml) were within the normal ranges. Only a small amount of
ascites and pleural effusion, with no evidence of an abnormal
image, were found on ultrasonography and computed tomography. In
addition, gastrointestinal endoscopy failed to demonstrate any
neoplasm other than chronic non-atrophic gastritis. An attendant
culdocentesis was performed and the ascitic fluid was found to be
positive for malignant cells consistent with metastatic
adenocarcinoma. Considering the prominent increase in CA125 and the
positive ascites, on December 23, 2012, laparoscopic exploration
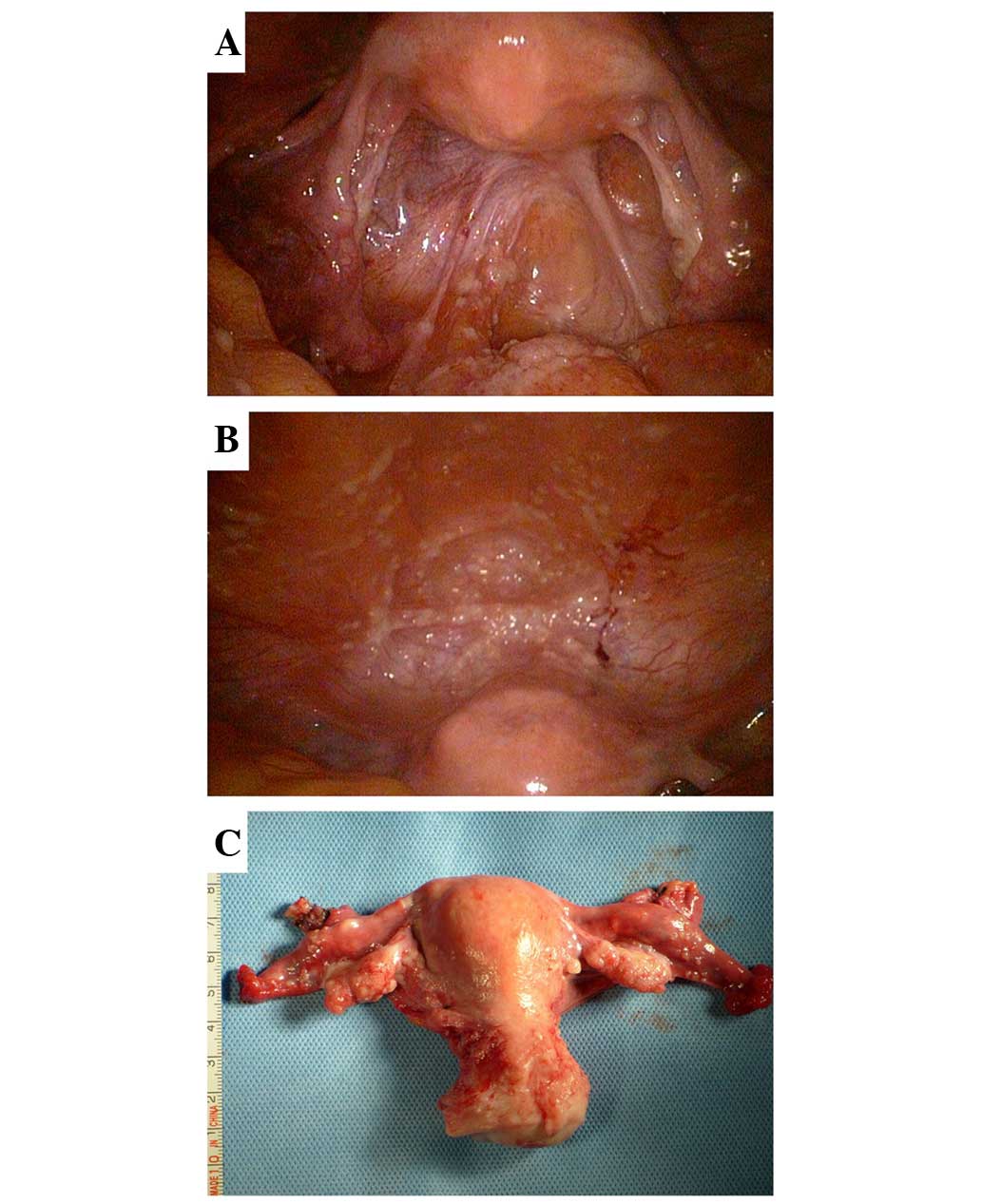
was recommended. It was observed that the bilateral ovaries
(10×20×30 mm) were slightly smaller than the normal size, with some
fine granular lesions on the surface. Similar diffuse micronodules
were scattered on the serosal surface of the uterus, fallopian
tubes, bladder, rectum, omentum, intestine and liver, as well as on
the diaphragm, and the abdominal and pelvic peritoneum. Moreover,
low-volume light green ascites were observed in the pelvic cavity
(Fig. 1).
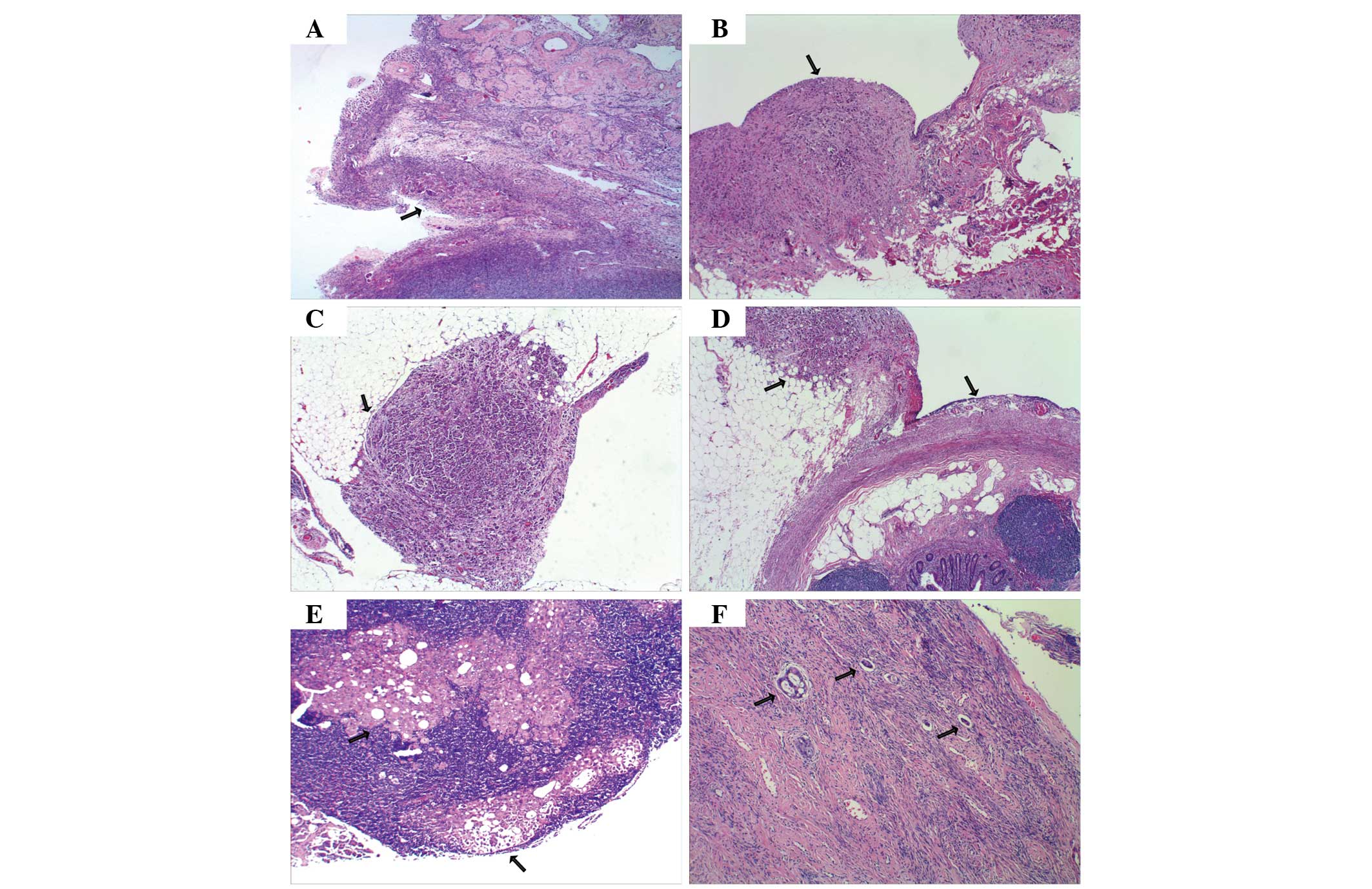
Post-operative histopathological evaluation of a 0.5
× 0.5 cm specimen removed from the peritoneum and ovary revealed a
poorly-differentiated serous adenocarcinoma, which infiltrated the
ovarian surface epithelium with no stromal invasion, and involved
the pelvic and abdominal peritoneum, the serosal surface of the
omentum and appendix, and the pelvic lymph nodes. Invasion into the
cervical vessels was of particular note (Fig. 2). Immunohistochemical staining of the
lesions presented positive for CA125, cytokeratin (CK)7, Ki-67,
CK19, E-cadherin, progesterone receptor (PR), EMA, estrogen
receptor (ER), Wilms' tumor protein (WT1) and p53, but negative for
CEA, CK5/6, AFP, CK20, human chorionic gonadotropin (HCG),
calretinin, thyroid transcription factor 1 (TTF1) and villin
(Fig. 3). The specimen resembled the
features of SOC derived from mullerian coelomic epithelium
differentiated from the peritoneal mesothelioma and metastases
(3,5–7). According
to the clinicopathological characteristics, the patient was
eventually diagnosed with EPSPC conforming to the GOG criteria
(2). A chemotherapy regimen
consisting of paclitaxel (135 mg/m2/24 h) plus cisplatin
(75 mg/m2) was administered intravenously at 3-week
intervals for 6 cycles. The serum CA125 level recovered to normal,
and to date, the patient has experienced disease-free survival and
is under follow-up.
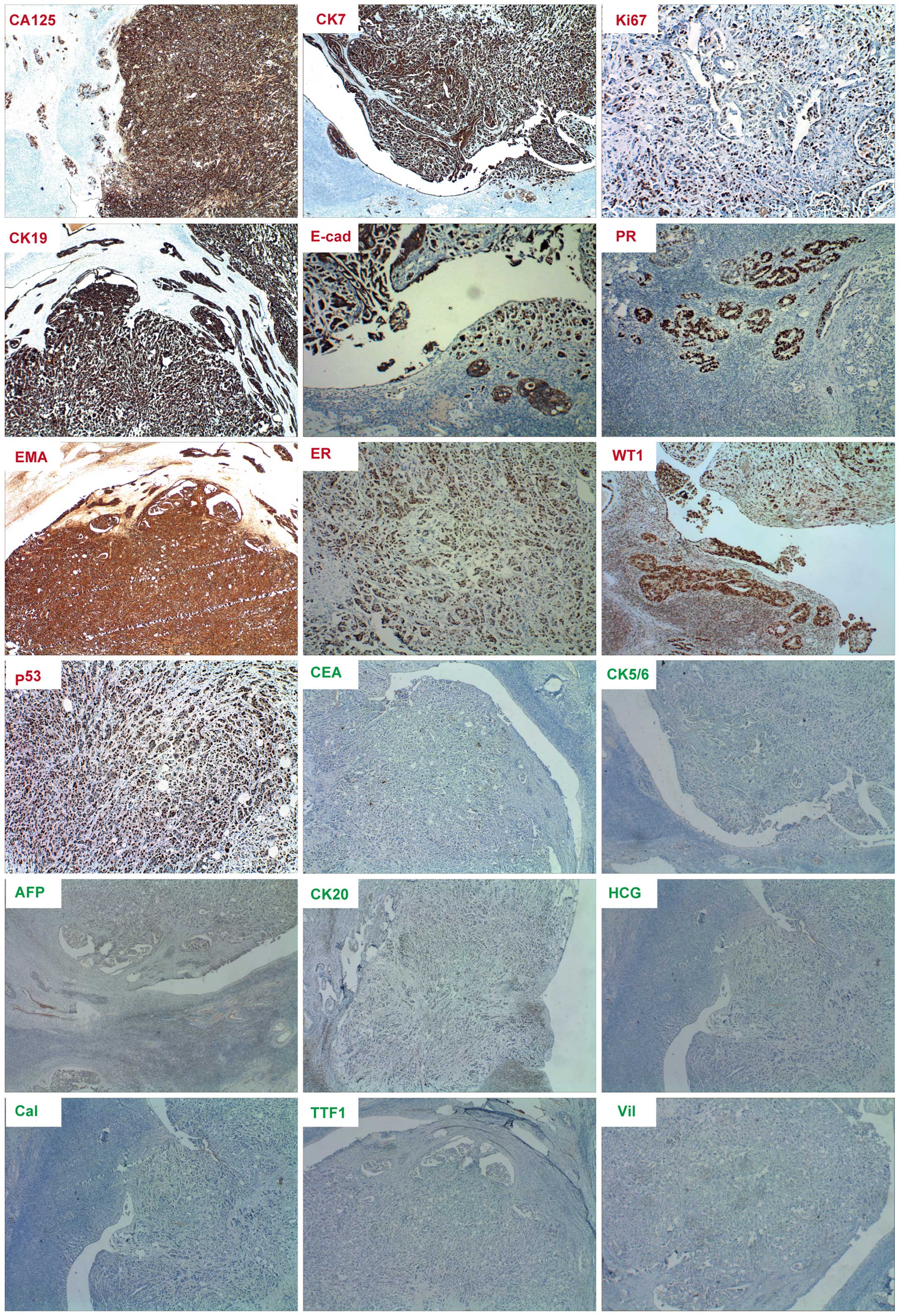 | Figure 3.Immumohistochemical staining of cancer
biomarkers in extraovarian peritoneal serous papillary carcinoma.
Positive staining (red lables) for cancer antigen 125 (CA125),
cytokeratin 7 (CK7), CK19, E-cadherin (E-cad), epithelial membrance
antigen (EMA), estrogen receptor (ER), progesterone receptor (PR),
Ki-67, tumor protein 53 (p53) and Wilms' tumor protein (WT1). The
results show negative staining for α-fetoprotein (AFP), calretinin
(Cal), carcinoembryonic antigen (CEA), CK5/6, CK20, human chorionic
gonadotropin (HCG), thyroid transcription factor 1 (TTF1) and
villin. Magnification for ER, Ki-67 and p53 is ×100, while
magnification for the remainder is ×40. |
The research was performed according to the
principles of the Declaration of Helsinki. Informed consent was
obtained from the patient and the study was approved by the Ethics
Committee of Yangpu Hospital.
Immunohistochemistry
Immunohistochemistry was performed using the
streptavidin-biotin complex method. Briefly, tissues were fixed
with 10% neutral-buffered formalin and paraffin-embedded, following
by cutting into 5-µm sections for immunohistochemical staining.
Following the hydration and antigen repair of tissues, the sections
were incubated overnight at 4°C with the following primary
antibodies: Mouse anti-CA125 monoclonal antibody (cat. no. M-0055;
Shanghai Long Island Biotec, Shanghai, China); mouse anti-CK7
monoclonal antibody (cat. no. GM7018; GeneTech, Co., Ltd.,
Shanghai, China); mouse anti-Ki-67 monoclonal antibody (cat. no.
GT2094; GeneTech, Co., Ltd.); mouse anti-CK19 monoclonal antibody
(cat. no. GT2255; GeneTech, Co., Ltd.); mouse anti-E-cadherin
monoclonal antibody (cat. no. GT2153; GeneTech, Co., Ltd.); rabbit
anti-PR monoclonal antibody (cat. no. F00392; Roche Diagnostics
(Shanghai) Ltd., Shanghai, China); mouse anti-EMA monoclonal
antibody (cat. no. GM0613; GeneTech, Co., Ltd.); rabbit anti-ER
monoclonal antibody (cat. no. F02583; Roche Diagnostics (Shanghai)
Ltd.); rabbit anti-WT1 polyclonal antibody (cat. no. R-0526;
Shanghai Long Island Biotec); mouse anti-p53 monoclonal antibody
(cat. no. R-0430; Shanghai Long Island Biotec); mouse anti-CEA
monoclonal antibody (cat. no. GT2108; GeneTech, Co., Ltd.); mouse
anti-CK5/6 monoclonal antibody (cat. no. GM7237; GeneTech, Co.,
Ltd.); rabbit anti-AFP polyclonal antibody (cat. no. GA0008;
GeneTech, Co., Ltd.); mouse anti-CK20 monoclonal antibody (cat. no.
GT2042; GeneTech, Co., Ltd.); mouse anti-HCG monoclonal antibody
(cat. no. R-0201; Shanghai Long Island Biotec); mouse
anti-calretinin monoclonal antibody (cat. no. GM3556; GeneTech,
Co., Ltd.); mouse anti-TTF1 monoclonal antibody (cat. no. GM3575;
GeneTech, Co., Ltd.); and rabbit anti-villin monoclonal antibody
(cat. no. R-0589; Shanghai Long Island Biotec). Subsequently, the
primary antibodies were detected using Dako EnVision Detection
Systems Peroxidase/DAB (Rabbit/Mouse) (Dako Japan Co., Ltd., Tokyo,
Japan). Negative controls were performed with normal mouse serum
(Dako Japan Co., Ltd.) or phosphate-buffered saline, and sections
were counterstained with hematoxylin.
Affymetrix OncoScan™ microarray
Sample DNA was extracted using a DNeasy Blood &
Tissue kit (cat. no. 69506; Qiagen GmBH, Hilden, Germany),
according to the manufacturer's instructions. A Nano Drop
spectrophotometer (cat. no. ND-1000; Thermo Fisher Scientific Inc.,
Waltham, MA, USA) and 1% agarose gel electrophoresis were used to
check the quantity and quality of the purified DNA. Sample DNA was
digested by the NspI restriction enzyme, and adaptors were
ligated to the fragment DNA to perform PCR amplification.
Amplification of the DNA was performed using the OncoScan™ FFPE
Assay kit (cat. no. 902293; Affymetrix, Santa Clara, CA, USA),
according to the manufacturer's protocol.
Amplified DNA was labeled and further fragmented
using an Affymetrix CytoScan HD Array Kit and Reagent Kit Bundle
(cat.no. 901835; Affymetrix), according to the manufacturer's
protocol. The Affymetrix OncoScan Assay User Manual (cat. no.
703038; Rev. 3; Affymetrix) was followed to obtain biotin-labeled
DNA. Hybridization buffers were prepared and array hybridization
was performed at 49°C in Hybridization Oven (cat. no. 00-0331-220V;
Affymetrix). After 16 h of hybridization, the arrays were washed in
a Fluidics Station (cat. no. 00-0079; Affymetrix) according to the
Affymetrix OncoScan™ Assay User Manual (cat. no. 703038; Rev. 3;
Affymetrix). Arrays were scanned using the GeneChip®
Scanner 3000 (cat. no. 00-00212; Affymetrix) and Command Console
Software 3.1 (Affymetrix) with default settings. Raw data that
passed quality control were further analyzed by
Affymetrix® OncoScan Analysis Suite (OncoScan Console
Software; Affymetrix).
Whole-exome sequencing
Primary tumor tissue consisted of blocks of fresh
frozen tissue acquired at the time of laparoscopy and then frozen
in liquid nitrogen. Genomic DNA was isolated from the primary tumor
tissue using the AllPrep DNA/RNA Mini kit (Qiagen). Genomic DNA was
isolated from blood to control for germline variants using the
DNeasy Blood and Tissue kit (Qiagen). The qualified genomic DNA
sample was randomly fragmented by Covaris and the size of the
library fragments was mainly distributed between 150 and 200 bp.
Next, adapters were ligated to each end of the resulting fragments.
The adapter-ligated templates were purified by the Agencourt AMPure
SPRI beads, and fragments with insert size of ~176 bp were excised.
Extracted DNA was amplified by ligation-mediated PCR (LM-PCR),
purified and hybridized to the SureSelect Biotiny lated RNA Library
(BAITS) for enrichment. Hybridized fragments were bound to the
strepavidin beads, whereas non-hybridized fragments were washed out
after 24 h. Captured LM-PCR products were subjected to a Agilent
2100 Bioanalyzer to estimate the magnitude of enrichment. Each
captured library was then loaded on a Hiseq2000 platform, and
high-throughput sequencing was performed for each captured library
to ensure that each sample met the desired average sequencing
depth. Raw image files were processed by Illumina basecalling
Software 1.7 (Illumina, San Diego, CA, USA) for basecalling with
default parameters, and the sequences of each individual were
generated as 90/100-bp pair-end reads.
The bioinformatics analysis began from the
sequencing data (raw data) generated from the Illumina pipeline.
Firstly, the adapter sequence in the raw data was removed, and low
quality reads that had too many Ns or a low base quality were
discarded. This step produced the ‘clean data’. Secondly, the
Burrows-Wheeler Aligner (BWA)-MEM algorithm in the BWA software
(https://sourceforge.net/projects/bio-bwa/files/) was
used to perform the alignment. BWA can provide results in binary
alignment/map (BAM) format files. The BAM format files were
required for certain processes, such as fixing mate information of
the alignment, adding read group information and removing duplicate
reads caused by PCR. After these processes, the final BAM files
used to do the variant calling were made ready. Single nucleotide
polymorphism (SNP) analysis was performed using the Short
Oligonucleotide Analysis Package for SNPs (release 1.03; http://soap.genomics.org.cn/soapsnp.html), Sequence
Alignment/Map tools (release 0.1.5–22; http://www.htslib.org/download/) or Genome Analysis
Toolkit (version 3.2; https://www.broadinstitute.org/gatk/). Following this,
filters were applied to obtain more confident variant results.
Subsequently, AnnoDB software (Bejing Genomics Institute, Beijing,
China) was used, which is in-house tool to annotate the confident
variant results. The final variants were fed into the downstream
advanced analysis pipeline. Quality control was present in the
whole pipeline, including for the clean data, the alignment and the
called variant.
Somatic single nucleotide variants (SNVs) occur in
any non-germ cell of the body following conception, such as in
those cells that initiate tumorigenesis. In the present study, the
Variant Detection in Massively Parallel Sequencing Data platform
(version 2.0; http://varscan.sourceforge.net/) was applied to
identify blood sample- and tumor sample-specific SNVs by
simultaneously comparing read counts, base quality and allele
frequency between the blood and tumor tissues. Somatic InDels are
mutations that occur due to small insertions or deletions of one or
a few nucleotides that occur in any non-germ cell of the body after
conception, such as those that initiate tumorigenesis. The strategy
for InDel analysis is as follows: In the sufficient covered sites,
an initial call is first made in the tumor sample, which is then
compared with the normal sample to find any evidence for the event;
if there is no evidence to support the InDel event in the normal
sample, this site will be considered a putative somatic InDel. The
probable impacts of the somatic mutations on an individual were
detected using the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT
(http://sift.bii.a-star.edu.sg/)
databases. Missense mutations predicted as ‘neutral’ by PolyPhen-2
were removed. The Kyoto Encyclopedia of Genes and Genomes pathway
database (http://www.genome.jp/kegg/pathway.html) was used for
pathway analysis.
Results
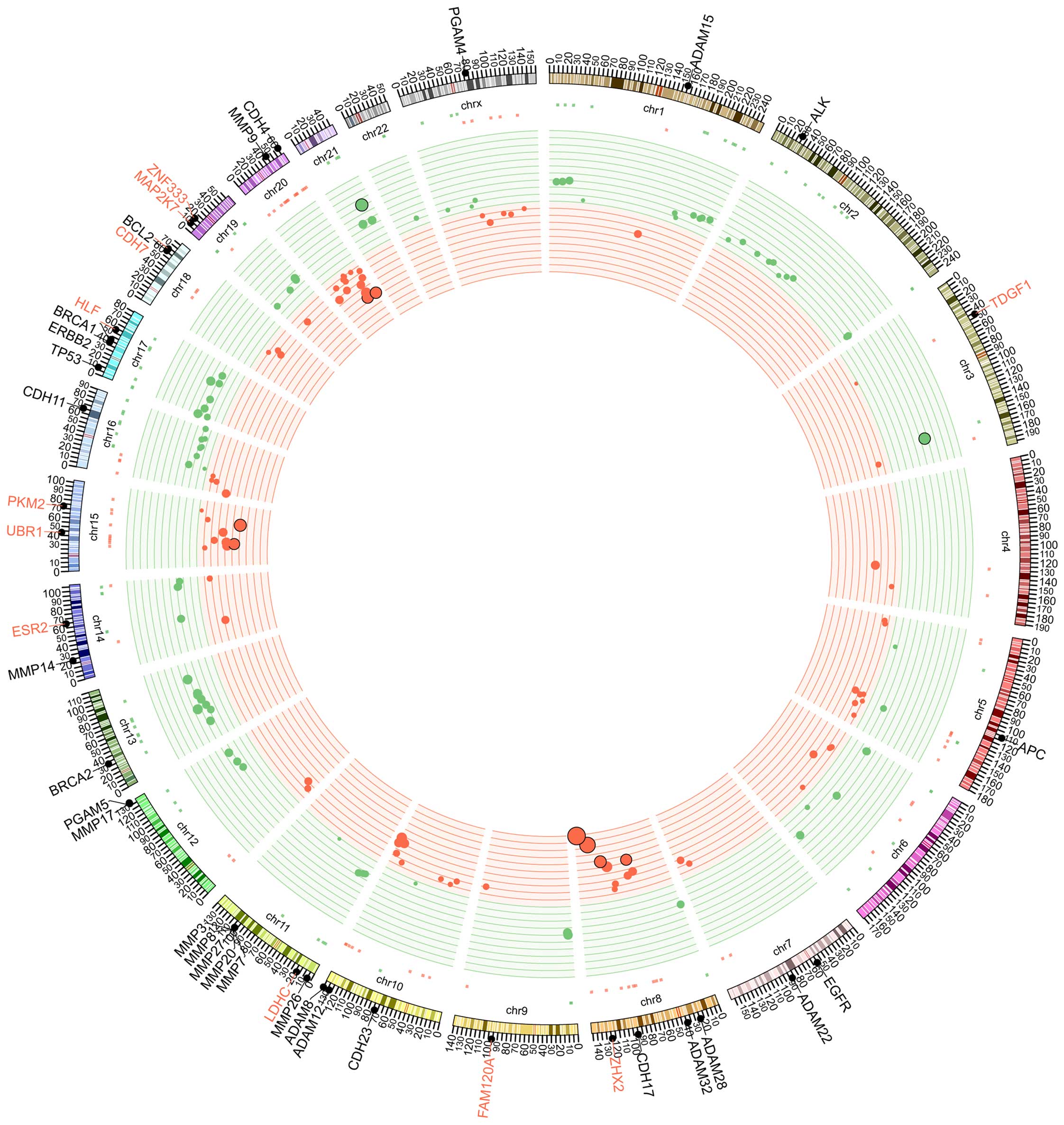
Copy number alteration in EPSPC
The CGH microarray was applied to analyze the copy
number variations by comparing tumor tissues and white blood cells
(WBCs). Unexpectedly, a number of large-scale copy number
alterations were identified in the tumor tissues (Fig. 4). Copy number gain occurred in a
variety of chromosomes, including 4q, 5q, 8q, 10q, 15q, 16p, 18q,
20p, 20q and Xq. In particular, chromosome 8 exhibited a high
number of gene amplifications in 8q. Copy number loss occurred in
numerous chromosomes, including chromosomes 1p, 1q, 2p, 6q, 8p, 9p,
11p, 12q, 13q, 16q, 17p, 17q, 19p, 19q, 21q, 22q and Xp. In
particular, large-scale copy number loss was found in chromosomes
2p, 13q, 16q, 17p and 17q.
Gene mutation in EPSPC
Whole-exome sequencing was applied to investigate
gene mutational spectra and insertion or deletion. A total of 14.6
billion bases of sequence were obtained (clean data/raw data,
92.13%) in tumor tissue, and a total of 15.2 billion bases of
sequence were obtained (clean data/raw data, 92.31%) in WBC DNA
(germline DNA). A total effective yield of 12,431 and 12,812 Mb
bases was found for tumor tissue DNA and WBC DNA, respectively. The
average read length was 89 bp for each. A total of 81,339 mutations
were identified in the germline DNA, including 8,062 missense
mutations and 9,563 synonymous mutations (data not shown). A total
of 11,286 short insertions or deletions were identified in the
germline DNA, including 187 frameshift insertions/deletions, 62
non-frameshift insertions and 77 non-frameshift deletions (data not
shown).
WBC DNA was analyzed to identify germline DNA
mutation (data not shown). The results showed that a few important
genes that are associated with cancer development were mutated,
such as BRCA1, DNA repair associated (BRCA1), which has 4 missense
mutation sites, including rs1799966 (Agt/Ggt), rs16942 (aAa/aGa),
rs16941 (gAa/gGa) and rs799917 (cCg/cTg). BRCA2 has 1 missense
mutation (rs169547; gTa/gCa). Tumor protein 53 (TP53) has 1
missense mutation (rs1042522, cCc/cGc). Erb-b2 receptor tyrosine
kinase 2 (ERBB2) has 1 missense mutation (rs1058808, Ccc/Gcc).
Additionally, oncogenes such as epidermal growth factor receptor
(EGFR) and KRAS proto-oncogene, GTPase (KRAS), and the tumor
suppressor gene, WNT signaling pathway regulator, also exhibited
mutations. Moreover, a number of cell migration-associated genes
were mutated, such as matrix metallopeptidase (MMPs) and ADAM
metallopeptidase domain-containing gene (ADAMs) family members
(Fig. 2).
Next, 165 somatic mutations were identified by
comparing tumor tissue DNA with WBC DNA, and the majority were
novel mutations (152/165), including 52 missense mutations, 15
synonymous mutations, 1 stop-gain mutation (immunoglobulin-like and
fibronectin type III domain containing 1), 7 short
insertions/deletions and 3 novel mutations, among others (data not
shown). In the functional prediction of these mutations, 70
potential driver mutations were screened out (data not shown), some
of which may be important for cancer metastasis and growth, such as
cadherin 7 (CDH7), family with sequence similarity 120A (FAM120A)
and ER 2 (ESR2). However, the majority of these mutations were
novel findings, which are unknown to cancer metastasis and require
further functional characterization.
Discussion
The present study investigated the copy number
variation and mutational spectra of a case with EPSPC, which may
provide novel insights into the course of disease evolution. Copy
number loss was previously reported in a study by Cass et al
(10), which screened for LOH at 39
chromosomal loci in 26 EPSPC and 37 SOC cases. Loci with LOH were
found in >30% of EPSPC cases in chromosomes 12p, 17p, 17q and
18q, as compared with large-scale loci with LOH in >30% of SOC
cases, located in chromosomes 4q, 5q, 6p, 6q, 9p, 9q, 12p, 12q,
13q, 15q, 16q, 17p, 17q, 18q, 19p, 19q, 22q and Xq. It appeared
that genomic stability was less frequent in EPSPC compared with
SOC. Huang et al similarly profiled 52 EPSPC and 33 SOC
cases for LOH at 22 microsatellite markers (11). LOH was commonly observed in the EPSPC
cases in loci 6q, 9p, 17p, 17q and Xq, while SOC cases commonly
harbored LOH in 1p, 7q, 11p, 17p and 17q. In the present study, the
patient showed a wide range of large-scale copy number alterations
across chromosomes. In accordance with the previous study by Huang
et al (11), this patient had
copy number loss in 6q, 9p, 17p and 17q. However, the patient also
exhibited many more copy number losses than previous studies, with
distinct copy number loss in 1q, 2p, 2q and Xp when compared with
SOC. Additionally, numerous copy number gains were found across the
chromosomes, including 4q, 5q, 8q, 10q, 15q, 16p, 18q, 20p, 20q and
Xq, implying a profound genomic rearrangement in the evolution of
EPSPC. Furthermore, the present study analyzed the genes located in
these gain regions and found certain notable indications, such as
in fibroblast growth factor (FGF)2 (located in 4q), FGF1 (located
in 5q) and FGF receptor (FGFR)2 (located in 10q), where the
amplification of these genes suggested that FGF-FGFR2 signaling may
be involved in tumor progression. The study also found other
cancer-related genes located in copy number gain regions, such as
telomerase reverse transcriptase in 5q, runt-related transcription
factor 1 in 8q, encoding mitogen-activated protein kinase kinase 5
in 15q and mitogen-activated protein kinase 3 in 16p. Collectively,
these copy number losses or gains may play essential roles in the
development of EPSPC.
There were numerous germline missense mutations in
the present case, suggesting that the cancer susceptibility genes,
including BRCA1, BRCA2, TP53, ERBB2, EGFR and KRAS, predisposed the
patient to the occurrence of the cancer. BRCA1 and BRCA2 had been
reported to be frequently mutated in EPSPC (12), and BRCA1 mutation may be associated
with a multifocal pathogenesis in such patients (13). BRCA1, BRCA2, TP53, ERBB2, EGFR and
KRAS also exhibit mutations in SOC (14–16), and
thus cannot be used as a unique feature to differentiate it from
EPSPC. However, unexpectedly, numerous MMP and ADAM family members
exhibited mutations in the present patient, which may have been a
predisposing factor for the high mobility of the cancer cells and
caused the wide distribution of cancer cells in the peritoneum.
Furthermore, 165 somatic mutations were found in the
present case; the majority of them were novel mutations, quite
different with the well-known driver oncogenes. For the functional
prediction of these mutations, 70 potential driver genes were
shown, some of which may be associated with tumor development.
CDH7, which is critical for epithelial-mesenchymal transition and
cell migration (17), may have an
impact on the metastasis of tumor cells. FAM120A may act in the
transport of mRNA in the cytoplasm, and is a vital component of
oxidative stress-induced survival signaling. FAM120A could act as
activator of src family kinases to phosphorylate and activate
phosphoinositide 3-kinase (18). ESR2
encodes ER β, which binds estrogens with an affinity similar to
that of ER α, and activates the expression of reporter genes
containing estrogen response elements in an estrogen-dependent
manner, as an ER to promote tumor growth (19). Collectively, profound genomic
rearrangements and somatic mutations may lead to the initiation and
development of EPSPC (Fig. 5).
However, the majority of these somatic mutations are novel and
require further functional characterization.
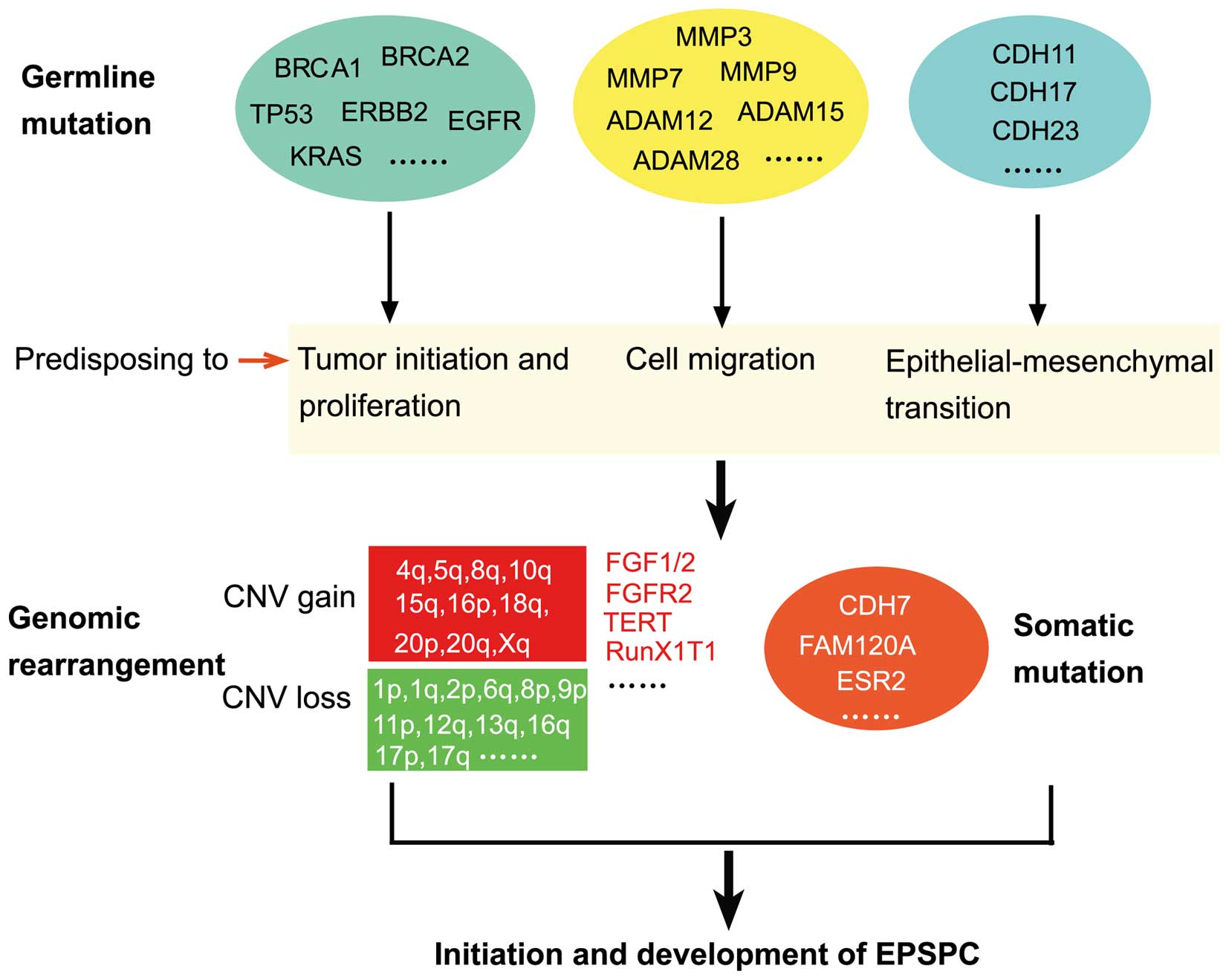 | Figure 5.Ideogram illustration of genomic
alterations that may be involved in the initiation and development
of EPSPC. Germline mutations may predispose an individual to the
disease, and then undergoing genomic rearrangement and somatic
mutation may lead to the occurrence and development of EPSPC.
BRCA1, BRCA1, DNA repair associated; TP53, tumor protein 53; ERBB2,
erb-b2 receptor tyrosine kinase 2; EGFR, epidermal growth factor
receptor; KRAS, KRAS proto-oncogene, GTPase; ADAM, ADAM
metallopeptidase domain-containing; MMP, matrix metalloproteinase;
CDH, cadherin; FGF, fibroblast growth factor; FGFR, FGF receptor;
TERT, telomerase reverse transcriptase; RUNX1T1, RUNX1
translocation partner 1; FAM120A, family with sequence similarity
120A; ESR2, estrogen receptor 2; CNV, copy number variation; EPSPC,
extraovarian peritoneal serous papillary carcinoma. |
In conclusion, the present study identified
large-scale copy number variations and the mutational spectra of
EPSPC. Germline DNA mutations appear to be much more profound than
somatic mutations, as numerous mutations were associated with cell
proliferation and migration, which suggested that the germline
mutations of this patient may predispose them to the occurrence of
EPSPC. Further studies are required to identify the functional
significance of these complex copy number variations and gene
mutations.
References
|
1
|
Swerdlow M: Mesothelioma of the pelvic
peritoneum resembling papillary cystadenocarcinoma of the ovary;
case report. Am J Obstet Gynecol. 77:197–200. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bloss JD, Liao SY, Buller RE, Manetta A,
Berman ML, McMeekin S, Bloss LP and DiSaia PJ: Extraovarian
peritoneal serous papillary carcinoma: A case-control retrospective
comparison to papillary adenocarcinoma of the ovary. Gynecol Oncol.
50:347–351. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhuyan P, Mahapatra S, Sethy S, Parida P
and Satpathy S: Extraovarian primary peritoneal papillary serous
carcinoma. Arch Gynecol Obstet. 281:561–564. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pentheroudakis G and Pavlidis N: Serous
papillary peritoneal carcinoma: Unknown primary tumour, ovarian
cancer counterpart or a distinct entity? A systematic review. Crit
Rev Oncol Hematol. 75:27–42. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goodman MT and Shvetsov YB: Rapidly
increasing incidence of papillary serous carcinoma of the
peritoneum in the United States: Fact or artifact? Int J Cancer.
124:2231–2235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dubeau L: The cell of origin of ovarian
epithelial tumours. Lancet Oncol. 9:1191–1197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grant DJ, Moorman PG, Akushevich L,
Palmieri RT, Bentley RC and Schildkraut JM: Primary peritoneal and
ovarian cancers: An epidemiological comparative analysis. Cancer
Causes Control. 21:991–998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao YN, Liu JX, Wang W, Li WF and Tang WS:
Comparison of primary extraovarian peritoneal serous papillary
carcinoma with stage III–IV ovarian papillary serous carcinoma.
Zhonghua Zhong Liu Za Zhi. 27:171–173. 2005.(In Chinese).
PubMed/NCBI
|
|
9
|
Kowalski LD, Kanbour AI, Price FV,
Finkelstein SD, Christopherson WA, Seski JC, Naus GJ, Burnham JA,
KanbourShakir A and Edwards RP: A case-matched molecular comparison
of extraovarian versus primary ovarian adenocarcinoma. Cancer.
79:1587–1594. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cass I, Baldwin RL, Fasylova E, Fields AL,
Klinger HP, Runowicz CD and Karlan BY: Allelotype of papillary
serous peritoneal carcinomas. Gynecol Oncol. 82:69–76. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang LW, Garrett AP, Schorge JO, Muto MG,
Bell DA, Welch WR, Berkowitz RS and Mok SC: Distinct allelic loss
patterns in papillary serous carcinoma of the peritoneum. Am J Clin
Pathol. 114:93–99. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bandera CA, Muto MG, Schorge JO, Berkowitz
RS, Rubin SC and Mok SC: BRCA1 gene mutations in women with
papillary serous carcinoma of the peritoneum. Obstet Gynecol.
92:596–600. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schorge JO, Muto MG, Welch WR, Bandera CA,
Rubin SC, Bell DA, Berkowitz RS and Mok SC: Molecular evidence for
multifocal papillary serous carcinoma of the peritoneum in patients
with germline BRCA1 mutations. J Natl Cancer Inst. 90:841–845.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McBride DJ, Etemadmoghadam D, Cooke SL,
Alsop K, George J, Butler A, Cho J, Galappaththige D, Greenman C,
Howarth KD, et al: Tandem duplication of chromosomal segments is
common in ovarian and breast cancer genomes. J Pathol. 227:446–455.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Castellarin M, Milne K, Zeng T, Tse K,
Mayo M, Zhao Y, Webb JR, Watson PH, Nelson BH and Holt RA: Clonal
evolution of high-grade serous ovarian carcinoma from primary to
recurrent disease. J Pathol. 229:515–524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bashashati A, Ha G, Tone A, Ding J,
Prentice LM, Roth A, Rosner J, Shumansky K, Kalloger S, Senz J, et
al: Distinct evolutionary trajectories of primary high-grade serous
ovarian cancers revealed through spatial mutational profiling. J
Pathol. 231:21–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Winklmeier A, ContrerasShannon V, Arndt S,
Melle C and Bosserhoff AK: Cadherin-7 interacts with melanoma
inhibitory activity protein and negatively modulates melanoma cell
migration. Cancer Sci. 100:261–268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanaka M, Sasaki K, Kamata R, Hoshino Y,
Yanagihara K and Sakai R: A novel RNA-binding protein,
Ossa/C9orf10, regulates activity of Src kinases to protect cells
from oxidative stress-induced apoptosis. Mol Cell Biol. 29:402–413.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leung YK, Mak P, Hassan S and Ho SM:
Estrogen receptor (ER)-beta isoforms: A key to understanding
ER-beta signaling. Proc Natl Acad Sci USA. 103:13162–13167. 2006.
View Article : Google Scholar : PubMed/NCBI
|