Introduction
T-acute lymphoblastic leukemia (T-ALL) is
characterised by the infiltration of bone marrow with immature
lymphoblasts that express T-cell immunophenotypic markers (1). T-ALL accounts for 10–15% of pediatric
and 25% of adult ALL (2). Various
genetic mutations have contributed to the pathogenesis of T-ALL.
Constitutive activation of Notch1, attributed to various mutations,
has been identified in over half of T-ALL patients (3,4). Notch1 is
a transmembrane protein frequently associated with various
oncogenic properties, including the promotion of cell survival,
angiogenesis and resistance to current chemotherapies; therefore,
it has attracted interest as a novel target for cancer therapy
(5,6).
γ-secretase is a membrane-embedded aspartyl protease responsible
for the cleavage of several transmembrane proteins, including Notch
and amyloid precursor protein (APP). The γ-secretase complex is
required for effective Notch signaling (7). Small molecule γ-secretase inhibitors
(GSIs) initially began clinical evaluation as a novel treatment for
Alzheimer's disease, since they block the accumulation of APP
(8). Subsequently, GSIs have been
clinically evaluated as Notch targeting anti-cancer agents.
Clinically evaluated GSIs include semagacestat (LY450139),
RO4929097, avagacestat (BMS-708163), PF-03084014 and 3-[(1r,
4s)-4-(4-chlorophenylsulfonyl)-4-(2,5-difluorophenyl) cyclohexyl]
propanoic acid (MK-0752) (data concerning these trials are
available at clinicaltrials.gov.). Initial results from preclinical
evaluations and clinical trials of GSIs have been disappointing,
due to the lack of anti-tumour activity, toxicity and resistance
(1,9,10).
However, recent phase I clinical trials with the novel GSI
PF-03084014, exhibited one complete response and several partial
responses in advanced cancers (11);
therefore, warranting continued clinical evaluation.
Combination therapies may overcome problems
associated with drug toxicity and resistance. The use of
combination regimes have significantly improved the outcome of
pediatric patients with T-ALL (12).
The tumour necrosis factor-related apoptosis-inducing ligand
(TRAIL)-receptor system has attracted immense attention as a
tumour-selective agent in terms of investment and clinical
evaluation. The TRAIL death receptor (DR) 4 and DR5 are exclusively
expressed on tumour cells (13).
Ligands, including TRAIL, which bind to TRAIL receptors, may
selectively activate the death signal in tumour cells (14). However as with numerous therapies,
resistance has been identified for this type of therapy; endogenous
low expression of DR4 and DR5 is responsible for inherent TRAIL
resistance in T-ALL patients (15).
Since, therapeutic agents that upregulate the expression of DR4 and
DR5 may restore sensitivity to TRAIL-induced apoptosis, the present
study investigated the potential of GSI and TRAIL combination
treatment to synergistically induce apoptosis in T-ALL cells by
restoring the DR-mediated pathway of apoptosis.
Materials and methods
Reagents
Reagents were purchased from Sigma-Aldrich (Poole,
UK), unless otherwise stated, and tissue culture vessels were
obtained from Greiner Bio-One GmbH (Frickenhausen, Germany).
Recombinant human TRAIL (amino acid, 114–281; catalog no., 616374)
and Caspase-8 Inhibitor z-IETD-FMK were purchased from Merck
Millipore (Nottingham, UK). TRAIL was supplied in a buffer
containing 500 mM NaCl, 10 mM Na2HPO4, 2.7 mM
KCl, 2 mM KH2PO4, 1 mM dithiothreitol (DTT),
≤10% glycerol (1.2 mg/ml), and stored in aliquots at −70°C.
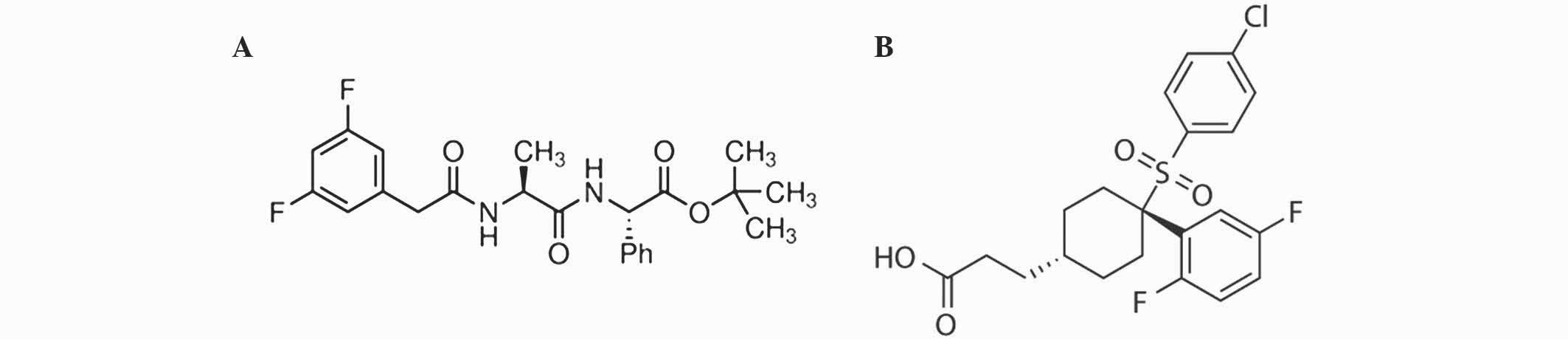
N-[(3,5-difluorophenyl) acetyl]-L-alanyl-2-phenyl]
glycine-1,1-dimethylethyl ester (DAPT; catalog no., D5942) was
purchased from Sigma-Aldrich. MK-0752 (catalog no., S2660) was
purchased from Selleckchem (Houston, TX, USA). Chemical structures
of DAPT and MK-0752 are shown in Fig.
1. Caspase inhibitors and MK-0752 were dissolved in dimethyl
sulfoxide (DMSO) and DAPT was dissolved in ethanol and stored at
−20°C. The final concentration of DMSO and ethanol did not exceed
0.1% (v/v) and 0.2% (v/v), respectively.
Cell culture
T-ALL Jurkat cells were obtained from
Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen und
Zellkulturen GmbH (Braunschweig, Germany). The cells were cultured
in RPMI-1640 containing GlutaMAX-1 supplemented with 10% fetal
bovine serum, 50 U/ml penicillin and 50 µg/ml streptomycin at 37°C
under 5% CO2. All cell culture materials were purchased
from Gibco® (Thermo Fisher Scientific, Inc., Waltham,
MA, USA).
Flow cytometric analysis of DNA
content
T-ALL cells were treated with TRAIL (20, 40, 100 and
200 ng/ml), DAPT (0, 10, 20 and 50 µM) or MK-0752 (50 µM), or a
combination of TRAIL and one GSI. The cells were also treated with
20 µM z-IETD-FMK in later experiments. Subsequent to 48 h, the
cells were pelleted by centrifugation at 800 × g and fixed with
ice-cold 70% ethanol with phosphate-buffered solution (PBS)
overnight at −20°C. Subsequently, the cells were treated with RNase
A (0.5 mg/ml) and stained with propidium iodide (0.15 mg/ml). DNA
content was measured using the BD Accuri C6 Flow Cytometer and
analysed with associated software (BD Biosciences, Inc., Franklin
Lakes, NJ, USA).
Western blotting
T-ALL cells were treated with 20 ng/ml TRAIL, 50 µM
DAPT or a combination of the two. Following treatment, the cells
were washed and re-suspended in PBS, lysed by addition of an equal
volume of 2X Laemmli buffer [1X = 30 mM Tris base (pH 6.8), 2%
sodium dodecyl sulfate, 10% glycerol] and briefly sonicated.
Protein content was measured using Pierce™ BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). DTT was added at a final
concentration of 50 mM following the BCA assay. Proteins were
denatured by boiling at 100°C for 5 min. Protein samples were
resolved on 4–20% Mini-PROTEAN® TGX™ Precast gels
(Bio-Rad Laboratories Ltd., Hemel Hempstead, UK) followed by
transfer to Immobilon-P PVDF membranes (EMD Millipore, Billerica,
MA, USA). The membranes were stained with 0.1% Ponceau S (w/v) in
5% acetic acid to ensure equal transfer. Only membranes exhibiting
equal loading and even transfer were used for further analysis. The
membranes were briefly washed in Tris-buffered saline (pH 7.7) and
0.05% Tween-20 (TBS-T) and blocked at room temperature in blocking
buffer [5% (w/v) dried milk dissolved in TBS-T]. After 1 h, the
membranes were probed with primary antibodies overnight at 4°C.
Rabbit monoclonal anti-cleaved Notch1 (Val1744; clone, D3B8;
catalog no., 4147; 1:500), rabbit monoclonal anti-x-linked
inhibitor of apoptosis (XIAP; clone, 3B6; catalog no., 2045;
1:1,000), rabbit polyclonal anti-BH3 interacting-domain death
agonist (Bid; catalog no., 2002; 1:1,000) and mouse monoclonal
anti-caspase-8 (clone, 1C12; catalog no., 9746; 1:1,000) antibodies
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). Mouse monoclonal anti-caspase-3 (clone, AM1.4.1-1B; catalog
no., AM39; 1:1,000) and anti-α-tubulin (clone, DM1A; catalog no.,
CP0; 1:2,500) antibodies were obtained from Merck Millipore.
Subsequently, the membranes were washed three times in TBS-T and
probed for 1 h at room temperature with horseradish
peroxidase-conjugated anti-mouse (cat. no. W4021) or anti-rabbit
(cat. no. W4011) secondary antibodies (1:1,000; Promega
Corporation, Madison, WI, USA) secondary antibodies. The membranes
were then exposed to Clarity™ ECL Western Blotting Substrate
(Bio-Rad Laboratories Ltd.) for 2 min and images were detected
using the Bio-Rad GelDoc XR system.
Flow cytometric analysis of DR5
T-ALL cells were treated with 50 µM DAPT. Following
treatment, the cells were washed in ice cold PBS and re-suspended
in PBS supplemented with 0.1% bovine serum albumin. DR5 cell
surface expression was analysed using fluorescein isothiocyanate
(FITC)-conjugated mouse monoclonal anti-TRAIL-R2 (clone, YM366;
catalog no., MAB10418; Merck Millipore) for DR5 analysis. FITC
conjugated IgG was used as an isotype control. The cells
(1×105) were incubated on ice with 10 µg/ml antibody for
45 min, washed in ice-cold PBS, re-suspended in FACS buffer and
analysed using the Accuri C6 Flow Cytometer. Values obtained for
the isotype control were subtracted from the anti-TRAIL-R2 values
and plotted accordingly.
Calculation of combination index
(CI)
Drug interactions were analysed using CalcuSyn 2.0
software (BIOSOFT, Cambridge, UK), as described by Bijnsdorp et
al (16). CIs were extrapolated
from drug cytotoxicity values derived from the quantification of
the sub-G1 (apoptotic) population of cells. CalcuSyn software
employs the Chou-Talalay method for drug combination, which is
based on the median-effect equation, derived from the mass-action
law principle (17). The resulting CI
values depict synergy (<1), additive effect (=1) and antagonism
(>1).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism software version 5.01 (GraphPad Software, Inc., La Jolla, CA,
USA). Data are presented as the mean ± standard error of the mean.
Comparisons between specific groups within data sets were assessed
using two-tailed Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Evaluation of GSI and TRAIL
combinations in human T-ALL cells
The effect of the GSI DAPT alone and in combination
with TRAIL in Jurkat T-ALL cells was determined by flow cytometric
evaluation of the pre-G1 peak. The present results revealed that
Jurkat cells are relatively resistant to DAPT up to concentrations
of 50 µM (Fig. 2A). Combining DAPT
with TRAIL (20–200 ng/ml) increased TRAIL-induced apoptosis in
Jurkat cells (Fig. 2B). CI was used
to determine synergism of DAPT and TRAIL combinations. Fig. 2C revealed that CI values ranged
between 0.018 and 0.165, which is indicative of clear synergy.
Furthermore, the second GSI MK-0752, which is currently under
clinical evaluation for various cancer types (details at clinicaltrials.gov), also significantly enhanced
TRAIL-induced apoptosis in T-ALL cells (Fig. 2D).
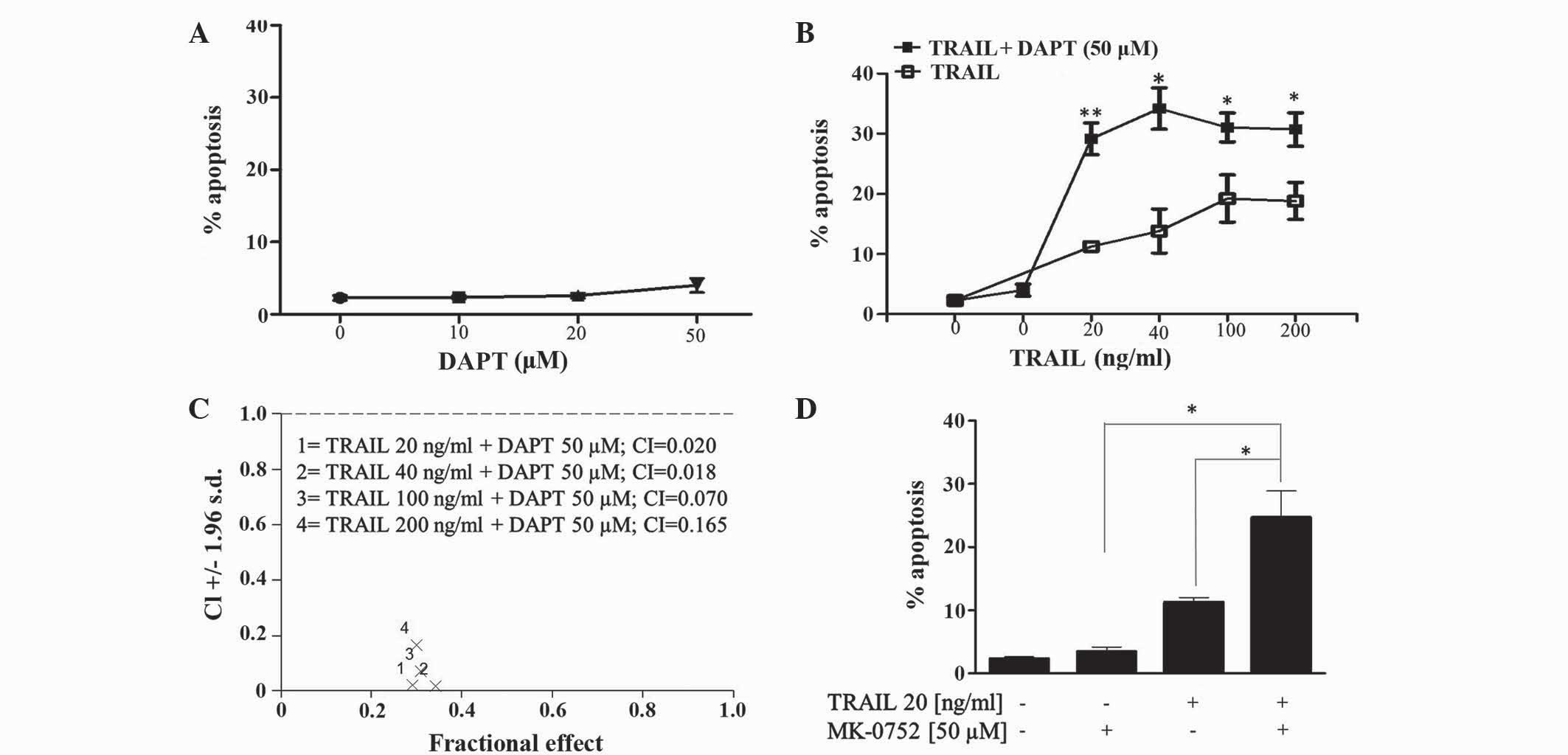 | Figure 2.Inhibition of γ-secretase enhances
TRAIL induced apoptosis in human T-cell acute lymphoblastic
leukemia Jurkat cells. Jurkat cells were treated with (A) DAPT (0,
10, 20 and 50 µM), (B and C) TRAIL (20, 40, 100 and 200 ng/ml),
DAPT (50 µM) or a combination of the two; and (D) TRAIL (20 ng/ml),
MK-0752 (50 µM) or a combination of the two for 48 h. The first
point on (B) refers to control vehicle-treated cells. The
percentage of apoptosis (pre-G1 cell population) was detected by
flow cytometric analysis of propidium iodide stained cells. Vehicle
control for (A-C), 0.2% (v/v) ethanol; (D), 0.1% (v/v) dimethyl
sulfoxide. Data shown are the mean ± standard error of the mean of
≥3 independent experiments. *P≤0.05, **P≤0.01. TRAIL, tumour
necrosis factor-related apoptosis-inducing ligand; DAPT,
N-[(3,5-difluorophenyl)
acetyl]-L-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester; MK-0752,
3-((1r, 4s)-4-(4-chlorophenylsulfonyl)-4-(2,5-difluorophenyl)
cyclohexyl) propanoic acid; CI, combination index. |
Effects of GSI and TRAIL combinations
on the expression of activated Notch and markers of apoptosis
Notch signaling is governed by the generation of the
intracellular domain of Notch1 (NCID) by γ-secretase, which
translocates to the nucleus and activates target genes. As
expected, inhibiting γ-secretase activity with DAPT (50 µM) reduced
the levels of NCID, as demonstrated by western blotting (Fig. 3A). TRAIL alone (20 ng/ml) did not
alter the expression of NCID. However, DAPT-mediated inhibition of
NCID formation was maintained in the presence of TRAIL.
DAPT-mediated enhancement of TRAIL-induced apoptosis appeared to
involve the extrinsic pathway, since activation of caspase-8 and
cleavage of the caspase-8 substrate Bid was observed by western
blotting (Fig. 3A). By contrast, the
effects on caspase-3 were minimum (Fig.
3A). In addition, DAPT potentiated TRAIL-induced reduction of
XIAP, which is a known inhibitor of the intrinsic and extrinsic
apoptosis pathway (Fig. 3A).
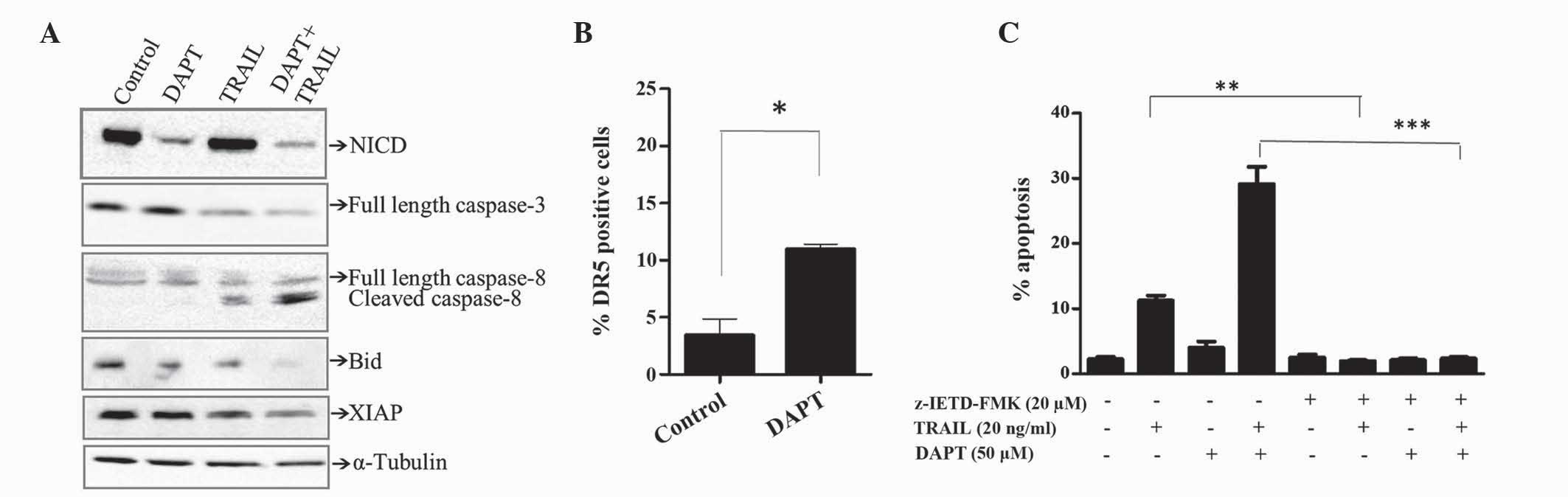 | Figure 3.Downregulation of XIAP together with
cleavage of caspase-8 and Bid are associated with DAPT/TRAIL
apoptotic synergy. Jurkat cells were treated for 48 h in the
presence of TRAIL (20 ng/ml), DAPT (50 µM) or a combination of the
two. (A) Expression of various proteins was determined by western
blotting. Equal loading was confirmed by α-tubulin. (B) Cell
surface expression of DR5 was determined using flow cytometric
analysis of cells stained with fluorescein
isothiocyanate-conjugated anti-TRAIL-R2. (C) Jurkat cells were
pre-treated for 1 h with the caspase-8 inhibitor z-IETD-FMK (20 µM)
or control [0.1% (v/v) dimethyl sulfoxide] then treated with TRAIL,
DAPT or a combination of the two. The percentage of apoptotic cells
was detected by flow cytometric analysis of propidium iodide
stained cells and quantification of the pre-G1 peak. Results are
representative of ≥3 independent experiments. *P<0.05, **P≤0.01,
***P≤0.001. XIAP, x-linked inhibitor of apoptosis; Bid, BH3
interacting-domain death agonist; TRAIL, tumour necrosis
factor-related apoptosis-inducing ligand; DAPT,
N-[(3,5-difluorophenyl)
acetyl]-L-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester; NCID,
intracellular domain of Notch1; DR, death receptor. |
TRAIL activates apoptosis by binding to its relevant
receptors, DR4 and DR5. Since DR5 is the predominant DR expressed
in Jurkat cells, the effect of DAPT on DR5 was determined by the
present study. As shown in Fig. 3B,
DAPT (50 µM) significantly increased the expression of DR5 in
Jurkat cells. Subsequently, the present study determined whether
caspase-8 was essential for TRAIL/DAPT apoptotic synergy. The
results obtained in Fig. 3C
demonstrate that apoptosis induced by TRAIL alone and in
combination with DAPT was completely inhibited by the caspase-8
inhibitor z-IETD-FMK, suggesting that caspase-dependent apoptosis
was mediated by the extrinsic apoptotic pathway.
Discussion
Initial enthusiasm afforded by oncologists towards
GSIs has dwindled, due to inherent tumour resistance to
Notch-targeting therapies. Resistance to established and emerging
chemotherapeutics, be it acquired or inherent, is an evolving
paradigm posing a significant challenge to clinicians. Once the
mechanism of resistance is established, rational therapeutic drug
combinations may subsequently be designed and pre-clinically
evaluated for potential therapeutic efficacy. T-ALL is a highly
aggressive hematopoietic malignancy prone to relapse and
resistance. Documented mechanisms of resistance include a low
expression of DRs (DR4 and DR5) (15)
and aberrations involving the T cell receptor, Notch1, HOXA
cluster, tyrosine kinases, cyclin-dependent kinase inhibitor 2A
locus and LIM domain only 2 proteins (2,18). The
present study targeted two such resistance mechanisms, low
expression of DR5 and Notch1, using two GSIs.
Various GSIs have been clinically evaluated as
bespoke anti-cancer agents targeting the γ-secretase-generated
Notch cleavage product NCID, which is associated with various
malignancies (19). Initial clinical
outcomes of GSI monotherapy with R04929097 fell short of clinical
expectations (20,21). Combination studies incorporating GSIs
with established anti-cancer treatments, including radiation;
Temsirolimus, an inhibitor of mammalian target of rapamycin
(20); capecitabine, a fluorouracil
pro-drug (22); bicalutamide, an
androgen antagonist; letrozole, a nonsteroidal aromatase inhibitor;
temozolomide, an alkylating agent; vismodegib, an inhibitor of the
hedgehog pathway and ABC transporters; tamoxifen, an antiestrogen;
erlotinib hydrochloride, an inhibitor of epidermal growth factor
receptor tyrosine kinase; gemcitabine hydrochloride, an
antimetabolite (23); vinblastine and
docetaxel, microtubule targeting agents; Cisplatin, a DNA targeting
agent; and cediranib maleate, a vascular endothelial growth factor
receptor-2 tyrosine kinase inhibitor (24) (details of not referenced drugs are
available from clinicaltrials.gov), have been evaluated or are
currently undergoing evaluation for therapeutic efficacy. For
certain trials with combination therapies, stable disease was the
best outcome (23–25), whilst other studies noted a partial
clinical response (22). Pre-clinical
research continues to aspire to identify suitable anti-cancer
agents to combine with GSIs, with the aim of further improving
clinical outcome.
As with GSIs, the TRAIL-DR ligand system primarily
presents as a promising tumour selective anti-cancer strategy.
However, TRAIL monotherapy has also succumbed to inevitable tumour
resistance. A previous study revealed that there was therapeutic
efficacy in combining GSI–I (Z-LLNle-CHO) with TRAIL in breast
carcinoma-derived cells (26);
Portanova et al (26)
demonstrated that inhibition of Notch may cooperate with TRAIL to
induce apoptosis in breast cancer cells.
The current study extends the pre-clinical
evaluation of TRAIL and GSIs as a possible therapy for the
treatment of T-ALL. The present results revealed a clear apoptotic
synergy in T-ALL cells when the GSI DAPT and TRAIL were
administered as a combination, as opposed to treatment with either
drug alone. A similar finding was observed when TRAIL was
administered with the novel GSI MK-0752. TRAIL interacts with its
receptors DR4 and DR5 and activates apoptosis via the extrinsic
apoptosis pathway. Specifically, binding of TRAIL to DR4 or DR5
leads to the formation of the death-inducing complex and
recruitment of the adaptor molecule Fas-associated protein with
death domain and subsequent activation of caspase-8 (27). The present study reports that the GSI
DAPT increases the levels of DR5 in Jurkat cells and re-sensitises
the cells to TRAIL-induced apoptosis. These findings are in
agreement with a previous study that revealed that GSI–I increased
the levels of DR4 and DR5 in breast cancer MDA-MB-231 cells
(26). By contrast to these findings,
ectopic expression of NCID sensitised hepatocellular carcinoma
cells to TRAIL-induced apoptosis by upregulating p53-dependent DR5
expression (28). However, a more
recent study demonstrated that Notch4, but not Notch1, confers
susceptibility to TRAIL-induced apoptosis in breast cancer cells
(29). Taken together, these findings
suggest that outcomes of TRAIL and Notch-based combination
strategies may be dependent on the cell type and the Notch isoform
expression profile, thus highlighting the requirement for
additional pre-clinical studies in other malignancies.
TRAIL induces apoptosis by binding to DR4 and DR5
and initiating the extrinsic pathway via activation of caspase-8.
In the present study, inhibition of caspase-8 using a selective
inhibitor completely blocked TRAIL and DAPT/TRAIL-induced
apoptosis; therefore confirming that DAPT enhances TRAIL-induced
apoptosis by activation of the extrinsic apoptotic pathway.
Caspase-8 initiates apoptosis by cleaving/activating caspase-3 or
by cleaving Bid into truncated (t)Bid. Bid was identified as the
preferred substrate of caspase-8 in vitro (30). It was previously reported that TRAIL
induces the formation of an active caspase-8/tBid complex on the
surface of the mitochondria, and thus activates the mitochondrial
apoptotic pathway (31). In the
present study, downregulation of Bid was more pronounced compared
with caspase-3 during DAPT/TRAIL apoptotic synergy, suggesting a
preference for Bid in the DAPT/TRAIL apoptotic pathway. A previous
study has revealed that Bid is required for stage II cleavage of
capase-3 (32). This suggests that
Bid is the preferred substrate of caspase-8 and is required for the
complete cleavage and activation of caspase-3. Bid was previously
defined as the mediator of apoptotic synergy induced by
TRAIL/etoposide combinations (33).
Furthermore, ectopic expression of Bid sensitised prostate cancer
cells to TRAIL-induced apoptosis (34). Taken together, these findings
highlight a role for Bid in apoptosis promoted by TRAIL-based
therapies. The anti-apoptotic protein XIAP inhibits mitochondrial
amplification of the extrinsic pathway by binding to and inhibiting
caspase-3, −7 and −9 (35). XIAP is
inhibited by Smac or Bid dependent cleavage (32,36).
Concomitant downregulation of XIAP during DAPT/TRAIL induced
apoptosis, as observed in the present study, may be a Bid-dependent
process. It is also possible that downregulation of XIAP may lead
to the activation of caspase-3 at later time points, resulting in
the subsequent amplification of the apoptotic signal by the
intrinsic pathway.
In the present study, DAPT alone caused a clear
decrease in NCID without inducing a significant amount of cell
death. This finding is in agreement with other studies
demonstrating that exposure of cells expressing Notch to GSI does
not necessary result in cell death (37). The present study demonstrates that
TRAIL maintains GSI-induced downregulation of NCID whilst
synergistically inducing apoptosis. Therefore, it may be suggested
that combining GSIs with other agents that trigger cell death
independently of Notch whilst maintaining GSI-induced
downregulation of Notch may prove to be a more successful method of
treating Notch driven malignancies. In conclusion, the present
study demonstrated that inhibition of γ-secretase presents a valid
strategy to overcome TRAIL resistance in T-ALL cells.
Acknowledgements
Dr Lisa M Greene and Dr Seema M Nathwani are
supported by a grant obtained from Children's Leukemia Research
Project (Dublin, Republic of Ireland).
Glossary
Abbreviations
Abbreviations:
|
T-ALL
|
T-cell acute lymphoblastic
leukemia
|
|
NCID
|
intracellular domain of Notch1
|
|
GSIs
|
γ-secretase inhibitors
|
|
DAPT
|
N-[(3,5-difluorophenyl)
acetyl]-L-alanyl-2-phenyl]glycine-1, 1-dimethylethyl ester
|
|
TRAIL
|
tumour necrosis factor-related
apoptosis inducing ligand
|
|
MK-0752
|
3-[(1r,
4s)-4-(4-chlorophenylsulfonyl)-4-(2,5-difluorophenyl) cyclohexyl]
propanoic acid
|
References
|
1
|
Ferrando AA: The role of NOTCH1 signaling
in T-ALL. Hematology Am Soc Hematol Educ Program. 353–361.
2009.PubMed/NCBI
|
|
2
|
Chiaretti S and Foà R: T-cell acute
lymphoblastic leukemia. Haematologica. 94:160–162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weng AP, Ferrando AA, Lee W, Morris JP IV,
Silverman LB, Sanchez-Irizarry C, Blacklow SC, Looks AT and Aster
JC: Activating mutations of NOTCH1 in human T cell acute
lymphoblastic leukemia. Science. 306:269–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haydu JE, De Keersmaecker K, Duff MK,
Paietta E, Racevskis J, Wiernik PH, Rowe JM and Ferrando A: An
activating intragenic deletion in NOTCH1 in human T-ALL. Blood.
119:5211–5214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Groth C and Fortini ME: Therapeutic
approaches to modulating Notch signaling: Current challenges and
future prospects. Semin Cell Dev Biol. 23:465–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takebe N, Nguyen D and Yang SX: Targeting
notch signaling pathway in cancer: Clinical development advances
and challenges. Pharmacol Ther. 141:140–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bray SJ: Notch signalling: A simple
pathway becomes complex. Nat Rev Mol Cell Biol. 7:678–689. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siemers ER, Quinn JF, Kaye J, Farlow MR,
Porsteinsson A, Tariot P, Zoulnouni P, Galvin JE, Holtzman DM,
Knopman DS, et al: Effects of a gamma-secretase inhibitor in a
randomized study of patients with Alzheimer disease. Neurology.
66:602–604. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deangelo DJ, Stone RM, Silverman LB, Stock
W, Attar EC, Fearen I, Dallob A, Matthews C, Stone J, Freedman SJ
and Aster J: A phase I clinical trial of the notch inhibitor
MK-0752 in patients with T-cell acute lymphoblastic
leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol (ASCO
Annual Meeting Proceedings). 24:65852006.
|
|
10
|
Kolb EA, Gorlick R, Keir ST, Maris JM,
Lock R, Carol H, Kurmasheva RT, Reynolds CP, Kang MH, Wu J, et al:
Initial testing (stage 1) by the pediatric preclinical testing
program of RO4929097, a γ-secretase inhibitor targeting notch
signaling. Pediatr Blood Cancer. 58:815–818. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Messersmith WA, Shapiro GI, Cleary JM,
Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds
JM, et al: A Phase I, dose-finding study in patients with advanced
solid malignancies of the oral γ-secretase inhibitor PF-03084014.
Clin Cancer Res. 21:60–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Litzow MR and Ferrando AA: How I treat
T-cell acute lymphoblastic leukemia in adults. Blood. 126:833–841.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Amarante-Mendes GP and Griffith TS:
Therapeutic applications of TRAIL receptor agonists in cancer and
beyond. Pharmacol Ther. 155:117–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hellwig CT and Rehm M: TRAIL signaling and
synergy mechanisms used in TRAIL-based combination therapies. Mol
Cancer Ther. 11:3–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Akahane K, Inukai T, Zhang X, Hirose K,
Kuroda I, Goi K, Honna H, Kagami K, Nakazawa S, Endo K, et al:
Resistance of T-cell acute lymphoblastic leukemia to tumor necrosis
factor-related apoptosis-inducing ligand-mediated apoptosis. Exp
Hematol. 38:885–895. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bijnsdorp IV, Giovannetti E and Peters GJ:
Analysis of drug interactions. Methods Mol Biol. 731:421–434. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Van Vlierberghe P and Ferrando A: The
molecular basis of T cell acute lymphoplastic leukemia. J Clin
Invest. 122:3398–3406. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Olsauskas-Kuprys R, Zlobin A and Osipo C:
Gamma secretase inhibitors of Notch signaling. Onco Targets Ther.
6:943–955. 2013.PubMed/NCBI
|
|
20
|
Diaz-Padilla I, Wilson MK, Clarke BA,
Hirte HW, Welch SA, Mackay HJ, Biagi JJ, Reedijk M, Weberpals JI,
Fleming GF, et al: A phase II study of single-agent RO4929097, a
gamma-secretase inhibitor of Notch signaling, in patients with
recurrent platinum-resistant epithelial ovarian cancer: A study of
the Princess Margaret, Chicago and California phase II consortia.
Gynecol Oncol. 137:216–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee SM, Moon J, Redman BG, Chidiac T,
Flaherty LE, Zha Y, Othus M, Ribas A, Sondak VK, Gajewski TF and
Margolin KA: Phase 2 study of RO4929097, a gamma-secretase
inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 121:432–440.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
LoConte NK, Razak AR, Ivy P, Tevaarwerk A,
Leverence R, Kolesar J, Siu L, Lubner SJ, Mulkerin DL, Schelman WR,
et al: A multicenter phase 1 study of γ -secretase inhibitor
RO4929097 in combination with capecitabine in refractory solid
tumors. Invest New Drugs. 33:169–176. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Richter S, Bedard PL, Chen EX, Clarke BA,
Tran B, Hotte SJ, Stathis A, Hirte HW, Razak AR, Reedijk M, et al:
A phase I study of the oral gamma secretase inhibitor R04929097 in
combination with gemcitabine in patients with advanced solid tumors
(PHL-078/CTEP 8575). Invest New Drugs. 32:243–249. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sahebjam S, Bedard PL, Castonguay V, Chen
Z, Reedijk M, Liu G, Cohen B, Zhang WJ, Clarke B, Zhang T, et al: A
phase I study of the combination of RO4929097 and cediranib in
patients with advanced solid tumours. Br J Cancer. 109:943–949.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Diaz-Padilla I, Hirte H, Oza AM, Clarke
BA, Cohen B, Reedjik M, Zhang T, Kamel-Reid S, Ivy SP, Hotte SJ, et
al: A phase Ib combination study of RO4929097, a gamma-secretase
inhibitor, and temsirolimus in patients with advanced solid tumors.
Invest New Drugs. 31:1182–1191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Portanova P, Notaro A, Pellerito O,
Sabella S, Giuliano M and Calvaruso G: Notch inhibition restores
TRAIL-mediated apoptosis via AP1-dependent upregulation of DR4 and
DR5 TRAIL receptors in MDA-MB-231 breast cancer cells. Int J Oncol.
43:121–130. 2013.PubMed/NCBI
|
|
27
|
Wang S and El-Deiry WS: TRAIL and
apoptosis induction by TNF-family death receptors. Oncogene.
22:8628–8633. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang C, Qi R, Li N, Wang Z, An H, Zhang Q,
Yu Y and Cao X: Notch1 signaling sensitizes tumor necrosis
factor-related apoptosis-inducing ligand-induced apoptosis in human
hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53
degradation and up-regulating p53-dependent DR5 expression. J Biol
Chem. 284:16183–16190. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Naik S, MacFarlane M and Sarin A: Notch4
signaling confers susceptibility to TRAIL-induced apoptosis in
breast cancer cells. J Cell Biochem. 116:1371–1380. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schug ZT, Gonzalvez F, Houtkooper RH, Vaz
FM and Gottlieb E: BID is cleaved by caspase-8 within a native
complex on the mitochondrial membrane. Cell Death Differ.
18:538–548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li S, Zhao Y, He X, Kim TH, Kuharsky DK,
Rabinowich H, Chen J, Du C and Yin XM: Relief of extrinsic pathway
inhibition by the Bid-dependent mitochondrial release of Smac in
Fas-mediated hepatocyte apoptosis. J Biol Chem. 277:26912–26920.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Broaddus VC, Dansen TB, Abayasiriwardana
KS, Wilson SM, Finch AJ, Swigart LB, Hunt AE and Evan GI: Bid
mediates apoptotic synergy between tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) and DNA damage. J Biol Chem.
280:12486–12493. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Orzechowska EJ, Kozlowska E, Czubaty A,
Kozlowski P, Staron K and Trzcinska-Danielewicz J: Controlled
delivery of BID protein fused with TAT peptide sensitizes cancer
cells to apoptosis. BMC Cancer. 14:7712014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wilkinson JC, Cepero E, Boise LH and
Duckett CS: Upstream regulatory role for XIAP in receptor-mediated
apoptosis. Mol Cell Biol. 24:7003–7014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang XD, Zhang XY, Gray CP, Nguyen T and
Hersey P: Tumor necrosis factor-related apoptosis-inducing
ligand-induced apoptosis of human melanoma is regulated by
smac/DIABLO release from mitochondria. Cancer Res. 61:7339–7348.
2001.PubMed/NCBI
|
|
37
|
Rao SS, O'Neil J, Liberator CD, Hardwick
JS, Dai X, Zhang T, Tyminski E, Yuan J, Kohl NE, Richon VM, et al:
Inhibition of NOTCH signaling by gamma secretase inhibitor engages
the RB pathway and elicits cell cycle exit in T-cell acute
lymphoblastic leukemia cells. Cancer Res. 69:3060–3068. 2009.
View Article : Google Scholar : PubMed/NCBI
|