Introduction
Prostate cancer is one of the most common malignant
tumors in men and the mortality rate has reached the second highest
ranking among all male cancers in the USA (1). Although the morbidity of prostate cancer
in China is far lower than that of western countries, the morbidity
has markedly increased in recent years, with an estimated incidence
of 10 cases/100,000 individuals in 2010 compared with 1.71
cases/100,000 individuals in 1993 (2). Early-stage prostate cancer requires
androgens for growth and thus responds well to androgen deprivation
therapy (3). However, the majority of
patients with androgen-dependent prostate cancer (ADPC) develop
androgen-independent prostate cancer (AIPC) subsequent to the
initiation of androgen deprivation. This indicates the next
proliferation stage of prostate cancer cells following the
remittent stage, such as continuous increase in prostate specific
antigen (PSA) levels, and bone metastasis occurs extremely easily.
At present, no curative therapy exists for this refractory
disease.
The AIPC transformation mechanism is the focus of
studies investigating prostate cancer. The primary reasons for ADPC
becoming AIPC are gene expression variation, signal pathway
abnormity, and dysregulation of proto-oncogenes, cancer suppressor
genes and growth factors (4). At
present, studies have identified a large number of relevant genes
and signal pathways of prostate cancer (5–7). However,
since the biological behavior of prostate cancer is extremely
complex, any of the aforementioned hypotheses cannot clarify the
pathogenetic mechanism of AIPC (6).
Therefore, identification of genes involved in the transition from
ADPC to AIPC is important to extend the current knowledge of AIPC
(7).
The human transcriptome comprises not only large
numbers of protein-coding messenger RNAs (mRNAs), but also a large
set of non-protein coding transcripts that have structural,
regulatory or unknown functions (8).
Noncoding RNAs are divided into long noncoding RNAs (>200
nucleoides) and short noncoding RNAs [20–30 nucleotides; microRNAs
(miRNAs)], according to their length. miRNAs are a class of small
noncoding RNAs that are implicated in numerous physiological and
pathological responses as post-transcriptional repressors of gene
expression. Mature miRNAs can specifically bind to the
3′-untranslated regions (3′-UTRs) of target cellular mRNA, in turn
triggering mRNA degradation or inhibition of translation (9). A previous study has reported that
miRNA-26a (miR-26a) had decreased expression in prostate cancer
(10). However, there is limited
knowledge about the role of miR-26a in prostate cancer.
Increased cytochrome c oxidase subunit II
(COX-2) expression has been linked to the initiation and
progression of human prostate cancer. TargetScan and luciferase
activity analysis have confirmed COX-2 as a target of miR-26a
(11), and COX-2 has been reported to
play a key role in the apoptosis of cancer cells (12).
In the present study, it was found that miR-26a was
significantly downregulated in prostate cancer tissues. Ectopic
expression of miR-26a may increase cell proliferation and inhibit
apoptosis. It was also found that miR-26a promotes apoptosis by
targeting COX-2.
Materials and methods
Cell culture and cell
transfection
The AIPC PC-3 cell line (purchased from the American
Type Culture Collection, Manassas, VA, USA) were maintained in
RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 2 mM L-glutamine (Sangong, Shanghai, China) and
25 mM HEPES (Sangong). miRNA mimics, inhibitors or scrambled
miRNA-control were synthesized from GenePharma (Shanghai, China)
products. pcDNA3.1-myc-COX-2 was purchased from Biogot Technology
Co., Ltd. (Nanjing, China). COX-2 siRNA was obtained from RiboBio
Co., Ltd. (Guangzhou, China) and the sequence was as follows:
Forward, 5′-GCUGGGAAGCCUUCUCUAA-3′; and reverse,
5′-TCGACCCUUCGGAAGAGAUU-3′. Transfections were performed using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
with miRNA mimics, inhibitors or scrambled control (RiboBio Co.,
Ltd.) for 24 h.
Plasmid constructs and luciferase
reporter assay
Targetscan was used to identify COX-2 as a potential
miR-26a target (http://www.targetscan.org/). To construct the
luciferase reporter vector, COX-2 3′-UTR and its flanking sequence
was amplified by polymerase chain reaction (PCR) using the
following primers: Forward, 5′-CGGGGTACCGAGTCATACTTGTGAAG-3′; and
reverse, 5′-GCACTCGAGCCTGTTTTTGTTTGATG-3′. The amplified fragment
was cloned into a PGL3 vector (Promega, Madison, WI, USA).
Similarly, a mutated COX-2 3′-UTR fragment, in which the miR-26a
binding site was mutated, was PCR-amplified using the following
primers; Forward, 5′-GTGGTTTCAACTTATATTATAAGAACG-3′; and reverse,
5′-GACGAAAAGACGTCAAAACTCATTT-3′. The PCR product was cloned into
the PGL3 vector. Luciferase activity assay was performed according
to the manufacturer's instructions (Promega, Madison, WI, USA).
Clinical samples
All prostate cancer and adjacent normal tissues
included in the present study were obtained from 32 patients with
prostate adenocarcinoma that were diagnosed by two pathologists at
the Department of Oncology, Urumqi General Hospital of Lanzhou
Military Command of the Chinese People's Liberation Army (Urumqi,
China). Medical history, transrectal ultrasound, computed
tomography and magnetic resonance imaging findings, and isotope
scanning of the skeleton were combined to decide the clinical
staging. In total, 12 patients that underwent radical
prostatectomy, did not have metastases and maintained extremely low
PSA levels (<0.2 ng/ml), with no relapse, were diagnosed with
ADPC, according to a previous study (13). In addition, 20 patients that presented
with increases in PSA levels or bone metastases were determined to
have advanced hormone-refractory prostate cancer. All experiments
were performed according to the principles of the Helsinki
Declaration. All patients enrolled in the present study provided
informed consent. The study was conducted with the approval of the
Ethical Committee of Urumqi General Hospital of Lanzhou Military
Command of the Chinese People's Liberation Army.
Cell proliferation assays
The methyl thiazolyl tetrazolium (MTT) assay, which
tests for cell proliferation and survival, was used in the present
study, as previously described (14).
Cells were incubated into 96-well culture plate, 5×103
cells in each well, to incubate for 72 h. In total, 50 µl MTT
solution was added to each well and incubated at 37°C for 4 h.
Following incubation, MTT was aspirated and 150 µl dimethyl
sulfoxide was added to each well to dissolve the formazan
precipitate. Subsequently, an ELISA reader (SM600; Utrao, Shanghai,
China) read the optical densities of the plates at 570 nm.
Cell apoptosis assay
An Annexin V-Fluos staining kit (Roche-Boehringer)
was used to detect early stages of apoptosis according with
previous report (9). The cells were
washed with phosphate-buffered saline (PBS) and stained according
to the manufacturer's protocol. Slides were mounted with Permafluor
mounting medium (Immunotech, Marseille, France) and viewed under a
fluorescence microscope (Axiophot; Olympus, Tokyo, Japan).
RNA extraction
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Reverse transcription-quantitative PCR
(RT-qPCR) analyses for miRNAs were performed using TaqMan miRNA
assays (Ambion; Thermo Fisher Scientific, Inc.) in an iQ5 Real-Time
PCR Detection system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). U6 small nuclear RNA was used as endogenous control for data
normalization. Relative expression was calculated using the
comparative threshold cycle (Ct) method. To identify apoptotic
nuclei, 4′,6-diamidino-2-phenylindole (Roche, Mannheim, Germany)
staining was performed according to the manufacturer's
protocol.
Western blot
The cells were washed with ice-cold PBS and then
lysed with protein lysate (Pierce, Rockford, IL, USA). After
centrifugation at 5,000 × g for 15 min at 4°C, the protein
concentration was measured with a bicinchoninic acid (BCA) protein
assay kit (Pierce). Subsequently, 50 µg aliquots of lysates were
loaded on a 10% sodium dodecyl sulfate-polyacrylamide gel and
transferred to a polyvinylidene difluoride membrane. The membranes
were blocked with 5% skimmed dry milk in Tris-buffered saline (pH
7.4) containing 0.05% Tween 20, and were incubated with Cox-2
antibodies (sc-514489; mouse monoclonal; 1:200 dilution; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and horseradish
peroxidase-conjugated mouse secondary antibodies (sc-2383; 1:5,000
dilution; Santa Cruz Biotechnology, Inc.), according to the
manufacturer's instructions. The protein of interest was visualized
using an enhanced chemiluminescence western blotting substrate
(Pierce) and the Chemidoc XRS Gel Documentation system (Bio-Rad
Laboratories, Inc.). Antibodies against glyceraldehyde 3-phosphate
dehydrogenase (G8795; mouse monoclonal; 1:5,000 dilution) were
obtained from Sigma-Aldrich (St. Louis, MO, USA).
Statistical analysis
The results are expressed as the mean ± standard
deviation of at least 3 separate experiments performed in
triplicate. The differences between groups were determined using
two-tailed Student's t-test, using SPSS software (version 22; IBM
Corporation, Armonk, NY, USA). P<0.05 was considered to indicate
a statistically significant difference. χ2 test or
Fisher's exact test was used to analyze the association between
miR-26a expression and the clinicopathological features.
Results
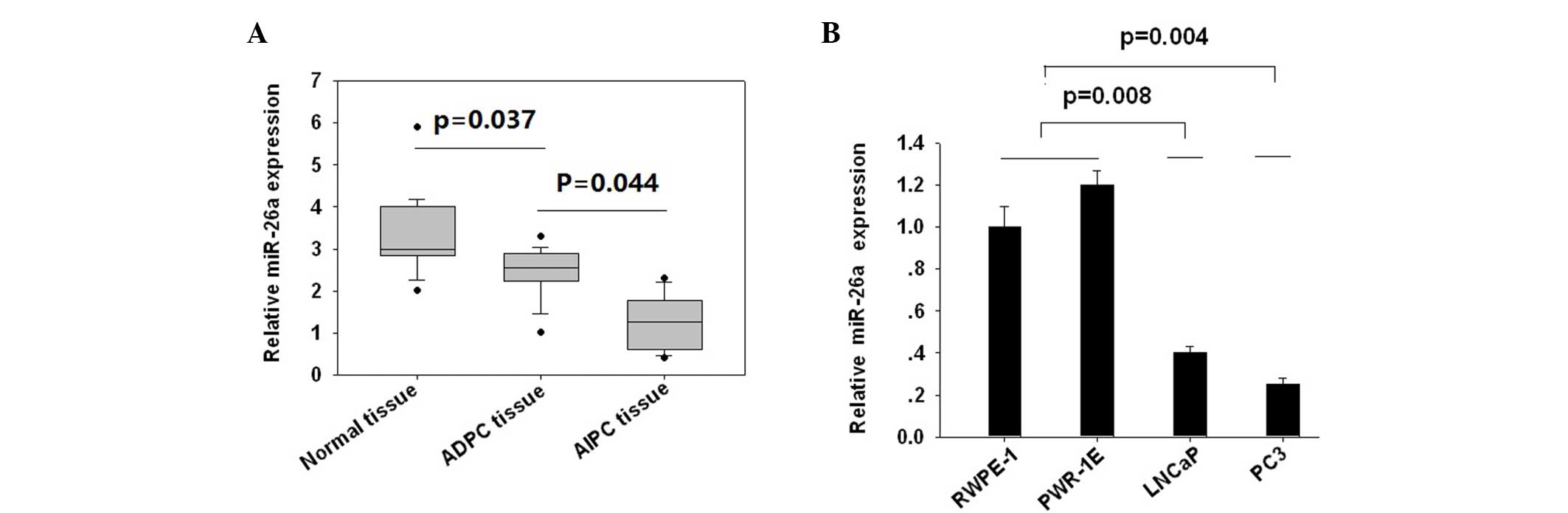
Expression of miR-26a was lower in
AIPC tissues compared with ADPC tissues
miR-26a expression was assessed in 12 AIPC tissues
and 20 ADPC cases, together with the matched adjacent non-tumor
prostate tissues. It was found that AIPC tissues showed the lowest
miR-26a expression, followed by ADPC groups (P=0.037; Fig. 1A), and the non-tumor tissues showed
significantly increased miR-26a expression compared with prostate
cancer tissues (P=0.044). To confirm these clinical findings, the
miR-26a expression using the ADPC LNCaP cell line, AIPC PC3 cell
line and the 2 normal prostate epithelial RWPE-1 and PWR-1E cell
lines. The expression of miR-26a was lowest in the PC3 cells,
followed by LNCaP cells (P=0.02). RWPE-1 and PWR-1E cells showed
higher miR-26a expression levels (Fig.
1B). These data indicated that miR-26a expression was decreased
in prostate cancer tissues and cell lines, with AIPC showing lower
miR-26a expression compared with ADPC.
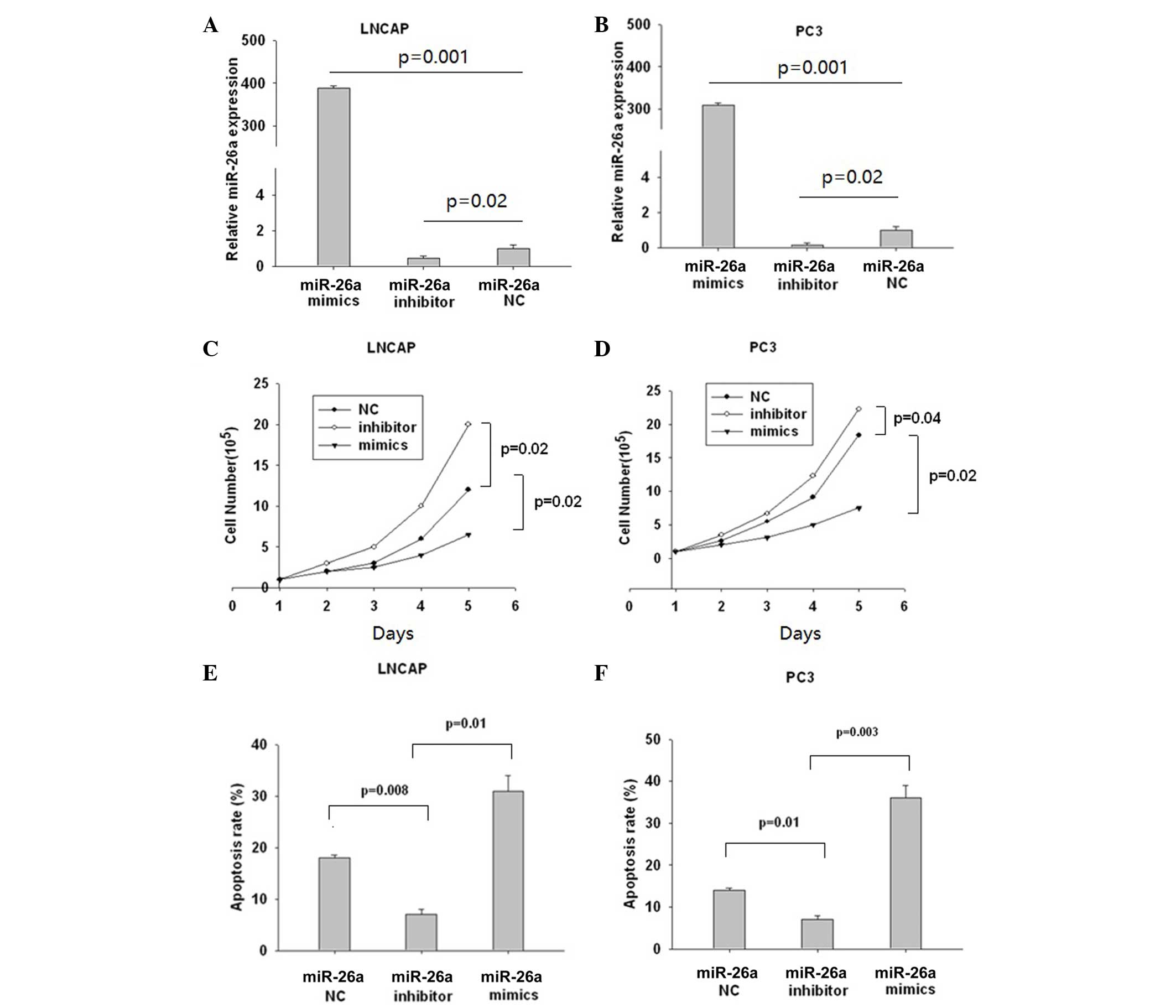
miR-26a negatively regulated prostate
cancer cell proliferation by enhancing cell apoptosis
LNCaP and PC3 cells were then transfected with
miR-26a mimics, inhibitors and control for 24 h and the efficiency
of transfection was shown in Fig. 2A and
B. miR-26a mimics significantly reduced the number of live
cells (P=0.02), while miR-26a inhibitors had little effect
(Fig. 2C). The same trend was also
observed in PC3 cells (Fig. 2D).
Subsequently, the cell apoptosis rates were determined using the
Annexin V-Fluos staining kit I in the LNCaP (Fig. 2E) and PC3 (Fig. 2F) cells miR-26a mimics significantly
enhanced the apoptosis rate (P=0.01), while miR-26a had the
opposite effect (P=0.008; Fig. 2E).
The same trend was observed in PC3 cells (Fig. 2F). These data indicated that miR-26a
performs an important role in apoptosis development.
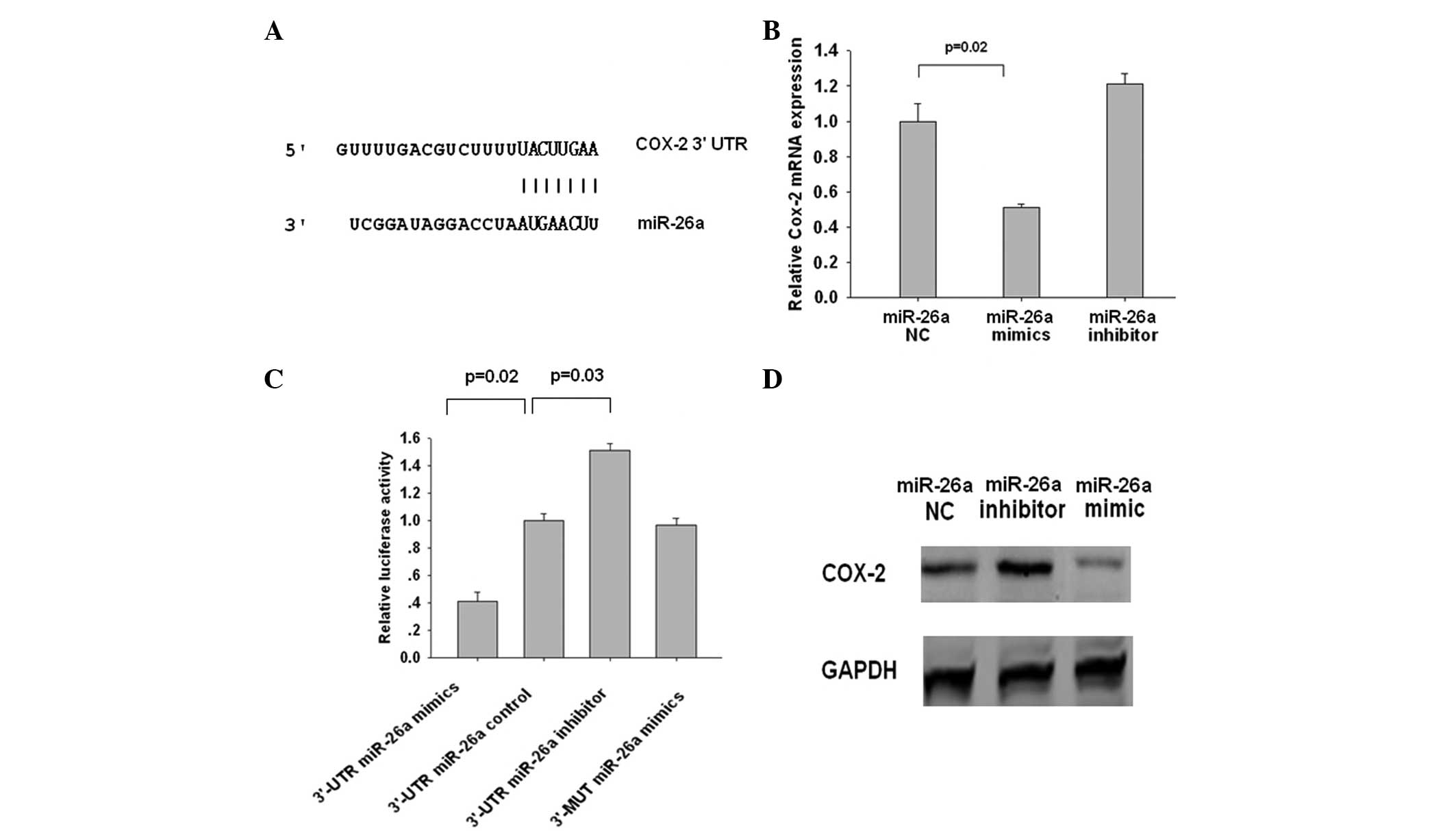
COX-2 is a miR-26a target gene in
prostate cancer cells
To determine the potential role of miR-26a in
mediating apoptosis, bioinformatic approaches identified potential
miR-26a targets, and miRNA target identification quality was
improved through the combined use of prediction programs. COX-2 was
identified as a putative miR-26a target gene (Fig. 3A). To directly address whether miR-26a
binds to the 3′-UTR of target mRNAs, luciferase reporter vectors
that contained the putative miR-26a binding sites within 3′-UTR and
the mutant 3′-UTR were generated. As shown in Fig. 3B, a marked reduction in luciferase
activity in cells transfected with miR-26a mimics and luciferase
report vectors was observed (P=0.02). By contrast, no change of
luciferase activity was observed in cells transfected with the
mutant 3′-UTR constructs.
miR-26a mediates COX-2 expression via
mRNA degradation
Measurement of the mRNA and protein levels of COX-2
in LNCaP and PC3 cells transfected with miR-26a mimics or
inhibitors revealed that overexpression (OV) of miR-26a resulted in
the downregulation of the mRNA and the protein levels of COX-2
(P=0.02), while miR-26a inhibitors had the opposite effect on the
target proteins (Fig. 3C and D).
These data suggest that COX-2 is a potential target of miR-26a in
prostate cancer cells and miR-26a mediates COX-2 expression via
mRNA degradation.
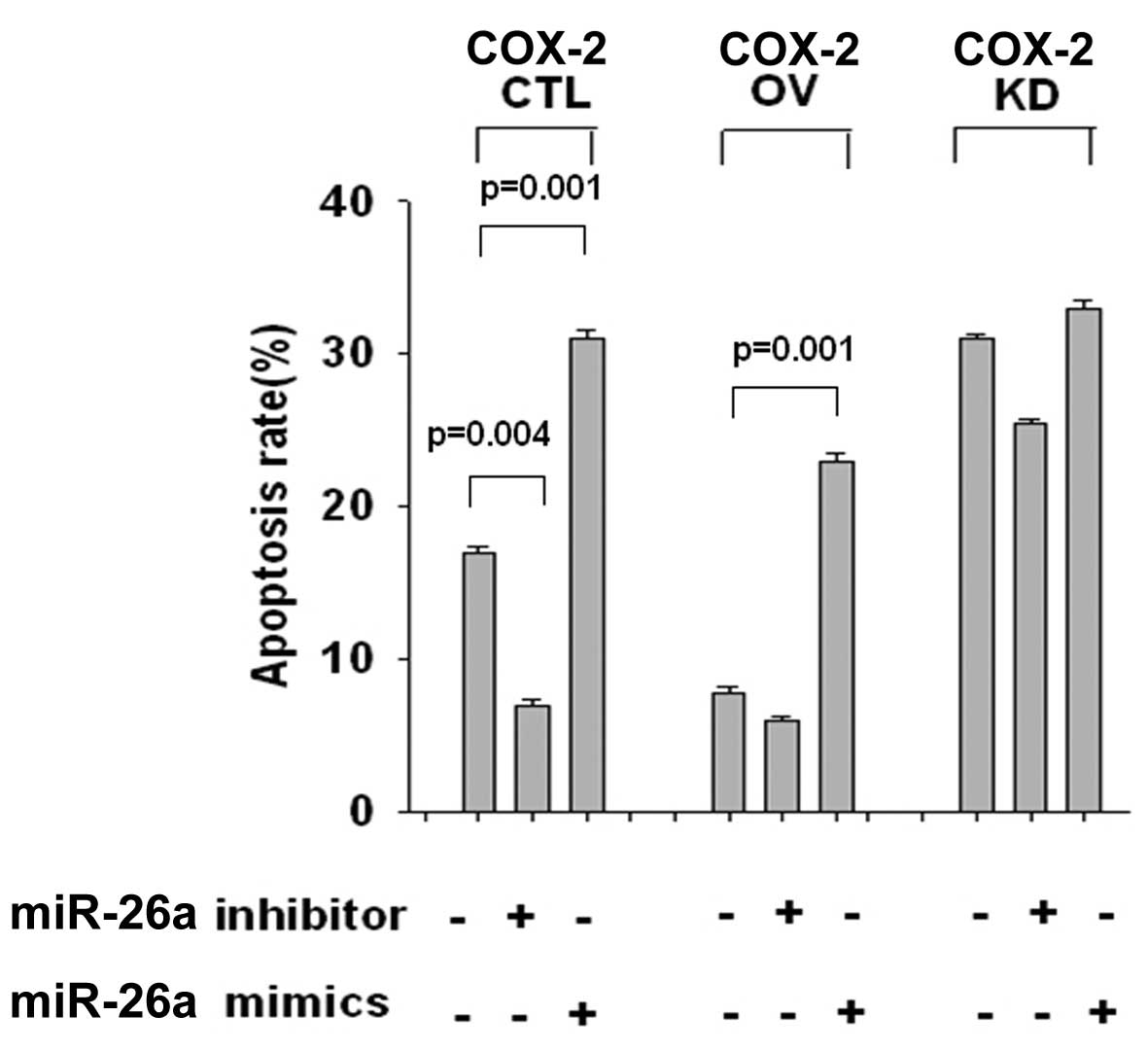
miR-26a increased cell apoptosis by
targeting COX-2 expression
COX-2 expression was associated with cancer cell
apoptosis; therefore, the subsequent investigations were designed
to explore whether miR-26a promotes apoptosis by suppressing COX-2.
Cells were transiently transfected with the pcDNA3.1-COX-2 OV and
siRNA for COX-2 knockdown (KD) plasmids, together with miR-26a
mimics, control or inhibitor. As shown in Fig. 4, apoptosis of LNCaP cells was markedly
reduced in the COX-2 OV group (P=0.001), and enhanced in the COX-2
KD group (P=0.003), indicating that COX-2 negatively regulated the
development of apoptosis. Cotransfection with miR-26a mimics
blocked the apoptosis reduction in the OV group (P=0.001), while in
the KD group, miR-26a mimics and miR-26a inhibitor had little
effect on the apoptosis rates. The same results were also observed
in PC3 cells (data not shown). These data suggested that miR-26a
upregulated apoptosis by suppressing COX-2 expression.
miR-26a expression and the association
with clinical features
The association between miR-26a mRNA expression and
clinical features of prostate cancer was analyzed in the high
(miR-26a/U6 mean value ≥2.1) and low (miR-26a/U6 mean value
<2.1) miR-26a expression groups, based on the results of RT-qPCR
analysis. As shown in Table I, low
miR-26a density was found in 14 cases of AIPC (70%), and only in 5
cases of ADPC (41.7%). In addition, intratumoral miR-26a density
was negatively associated with the Gleason score (P=0.02) and T
stage (P=0.04). These data indicated that patients with lower
miR-26a density had an evidently poor prognosis (Table I).
 | Table I.Association between miR-26a expression
and clinicopathological variables of patients with prostate
cancer. |
Table I.
Association between miR-26a expression
and clinicopathological variables of patients with prostate
cancer.
|
|
| miR-26a
expression |
|
|---|
|
|
|
|
|
|---|
| Variables | Cases | Low | High | P-value |
|---|
| Gleason score |
|
|
| 0.02 |
| ≤7 | 13 | 2 | 11 |
|
|
>7 | 19 | 11 | 8 |
|
| T stage |
|
|
| 0.04 |
| I+II | 11 | 5 | 6 |
|
|
III+IV | 21 | 17 | 4 |
|
| PSA dependence |
|
|
| 0.04 |
| AIPC | 20 | 14 | 6 |
|
| ADPC | 12 | 5 | 7 |
|
Discussion
The expression level of miR-26a has been found to be
downregulated in various types of cancers, and the miRNA was
demonstrated to be a tumor suppressor that inhibits cancer cell
proliferation by targeting various key proteins, such as EZH2
(15) and MCL-2 (16). By contrast, miR-26a was reported to
facilitate carcinogenesis through the inhibition of phosphatase and
tensin homolog (17). These studies
indicated that miR-26 may play a different role in different
tumors.
For prostate cancer, previous studies have
demonstrated that miR-26a expression was lower in tumor tissues
compared with their normal controls, which was also found in
prostate cancer cells compared with normal cells (18). Consistent with these studies, the
present study also found that the expression of miR-26a was
downregulated in prostate cancer tissues and cell lines. Notably,
miR-26a expression was significantly lower in the AIPC group
compared with the ADPC group. Furthermore, a significant
association was also observed between miR-26a expression and
increased Gleason scores (P=0.02), and between miR-26a expression
and tumor stage (P=0.04), indicating that the low miR-26a level was
predictive of a poor prognosis.
Cell based assay indicated that miR-26a OV resulted
in inhibition of tumor cell proliferation. In addition, the present
study, to the best of our knowledge, is the first to demonstrate
that negative regulatory function of miR-26a against COX-2 mediated
apoptosis promotion was the main reason for tumor inhibition. COX-2
expression is suggested to be triggered by a variety of cytokines,
and was shown to be a negative regulator of apoptosis (19). Accumulating evidence from experiments
and clinical trials indicate that COX-2 plays a role in human
carcinogenesis by inhibiting apoptosis, and COX-2 inhibitors were
demonstrated to be able to arrest prostate cancer growth (20,21). In
the present study, apoptosis was markedly reduced in miR-26a
inhibitor treatment, and then cotransfection si-COX-2 blocked this
apoptosis reduction, suggesting that miR-26a-regulated cell
apoptosis was dependent on COX-2.
Several miRNAs were demonstrated to modulate cell
function through targeting COX-2. Wu et al (9) showed that miR-146a enhances
Helicobacter pylori-induced cell apoptosis in human gastric
cancer epithelial cells by targeting COX-2. Hao et al
(22) demonstrated that enforced
expression of miR-101 inhibits prostate cancer cell growth by
modulating the COX-2 pathway. miR-16 has also been identified as a
potential tumor regulator by targeting COX-2 (23). In addition to the present findings,
these studies provide a novel cancer therapy based on miRNAs by
directly inhibiting COX-2 expression.
The present study may assist in our understanding of
the miR-26a regulation of AIPC formation in order to provide
insight into the unique character of prostate cancer development.
Furthermore, AIPC-related miR-26a could become a novel diagnostic
or therapeutic tool in prostate cancer. The main limitation of the
present study was that the small study and so further studies on a
greater number of patients are ongoing to aid the generalizability
of these conclusions.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ren SC, Chen R and Sun YH: Prostate cancer
research in China. Asian J Androl. 15:350–353. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sooriakumaran P, Nyberg T, Akre O,
Haendler L, Heus I, Olsson M, Carlsson S, Roobol MJ, Steineck G and
Wiklund P: Comparative effectiveness of radical prostatectomy and
radiotherapy in prostate cancer: Observational study of mortality
outcomes. BMJ. 348:g15022014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kahn B, Collazo J and Kyprianou N:
Androgen receptor as a driver of therapeutic resistance in advanced
prostate cancer. Int J Biol Sci. 10:588–595. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shi X, Gipp J, Dries M and Bushman W:
Prostate progenitor cells proliferate in response to castration.
Stem Cell Res. 13:154–163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ianni M, Porcellini E, Carbone I,
Potenzoni M, Pieri AM, Pastizzaro CD, Benecchi L and Licastro F:
Genetic factors regulating inlammation and DNA methylation
associated with prostate cancer. Prostate Cancer Prostatic Dis.
16:56–61. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu JB, Dai CM, Su XY, Cao L, Qin R and
Kong QB: Gene microarray assessment of multiple genes and signal
pathways involved in androgen-dependent prostate cancer becoming
androgen independent. Asian Pac J Cancer Prev. 15:9791–9795. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sullenger BA and Nair S: From the RNA
world to the clinic. Science. 352:1417–1420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu K, Yang L, Li C, Zhu CH, Wang X, Yao Y
and Jia YJ: MicroRNA-146a enhances Helicobacter pylori induced cell
apoptosis in human gastric cancer epithelial cells. Asian Pac J
Cancer Prev. 15:5583–5586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao S, Ye X, Xiao L, Lian X, Feng Y, Li F
and Li L: MiR-26a inhibits prostate cancer progression by
repression of Wnt5a. Tumor Biol. 35:9725–9733. 2014. View Article : Google Scholar
|
|
11
|
Shao Y, Li P, Zhu ST, Yue JP, Ji XJ, Ma D,
Wang L, Wang YJ, Zong Y, Wu YD and Zhang ST: MiR-26a and miR-144
inhibit proliferation and metastasis of esophageal squamous cell
cancer by inhibiting cyclooxygenase-2. Oncotarget. 7:15173–15186.
2016.PubMed/NCBI
|
|
12
|
Hickman OJ, Smith RA, Dasgupta P, Rao SN,
Nayak S, Sreenivasan S, Vyakarnam A and Galustian C: Expression of
two WFDC1/ps20 isoforms in prostate stromal cells induces paracrine
apoptosis through regulation of PTGS2/COX-2. Br J Cancer.
114:1235–1242. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jia RP, Xu LW, Su Q, Zhao JH, Li WC, Wang
F and Xu Z: Cyclooxygenase-2 expression is dependent upon epidermal
growth factor receptor expression or activation in androgen
independent prostate cancer. Asian J Androl. 10:758–764. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang D, Zhu X, Cui C, Dong M, Jiang H, Li
Z, Liu Z, Zhu W and Wang JG: Discovery of novel acetohydroxyacid
synthase inhibitors as active agents against Mycobacterium
tuberculosis by virtual screening and bioassay. J Chem Inf Model.
53:343–353. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang B, Liu XX, He JR, Zhou CX, Guo M, He
M, Li MF, Chen GQ and Zhao Q: Pathologically decreased miR-26a
antagonizes apoptosis and facilitates carcinogenesis by targeting
MTDH and EZH2 in breast cancer. Carcinogenesis. 32:2–9. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao J, Li L, Wu M, Liu M and Xie X, Guo J,
Tang H and Xie X: MiR-26a inhibits proliferation and migration of
breast cancer through repression of MCL-1. PLoS One. 8:e651382013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu B, Wu X, Liu B, Wang C, Liu Y, Zhou Q
and Xu K: MiR-26a enhances metastasis potential of lung cancer
cells via AKT pathway by targeting PTEN. Biochim Biophys Acta.
1822:1692–1704. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kato M, Goto Y, Matsushita R, Kurozumi A,
Fukumoto I, Nishikawa R, Sakamoto S, Enokida H, Nakagawa M,
Ichikawa T and Seki N: MicroRNA-26a/b directly regulate La-related
protein 1 and inhibit cancer cell invasion in prostate cancer. Int
J Oncol. 47:710–718. 2015.PubMed/NCBI
|
|
19
|
Sun WH, Zhu F, Chen GS, Su H, Luo C, Zhao
QS, Zhang Y, Shao Y, Sun J, Zhou SM, et al: Blockade of
cholecystokinin-2 receptor and cyclooxygenase-2 synergistically
induces cell apoptosis and inhibits the proliferation of human
gastric cancer cells in vitro. Cancer Lett. 263:302–311. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bieniek J, Childress C, Swatski MD and
Yang W: COX-2 inhibitors arrest prostate cancer cell cycle
progression by down-regulation of kinetochore/centromere proteins.
Prostate. 74:999–1011. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Walther U, Emmrich K, Ramer R, Mittag N
and Hinz B: Lovastatin lactone elicits human lung cancer cell
apoptosis via a COX-2/PPARγ-dependent pathway. Oncotarget.
7:10345–10362. 2016.PubMed/NCBI
|
|
22
|
Hao Y, Gu X, Zhao Y, Greene S, Sha W,
Smoot DT, Califano J, Wu TC and Pang X: Enforced expression of
miR-101 inhibits prostate cancer cell growth by modulating the
COX-2 pathway in vivo. Cancer Prev Res (Phila). 4:1073–1083. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Young LE, Moore AE, Sokol L, Meisner-Kober
N and Dixon DA: The mRNA stability factor HuR inhibits microRNA-16
targeting of COX-2. Mol Cancer Res. 10:167–180. 2012. View Article : Google Scholar : PubMed/NCBI
|